
ABOUT AUTHOR
SHRIKANT GODIYAL
Deputy Manager- Regulatory Affairs,
Tirupati Group, Poanta Sahib, Himachal Pradesh-173025, India
ABSTRACT
Expression of Interest is an Application which is submitted for Product Evaluation to the Expert Review Panel Prequalification Team World Health Organization (WHO) which evaluates the application along with the supporting documents and ensures that the Active Pharmaceutical Ingredients (API’s) and Finished Pharmaceutical Products (FPPs) are safe to use and all the studies w.r.t the safety and efficacy of the product had been completed.
INTRODUCTION:
World Health Organization come into existence after configuration of Constitution on 7th April 1948, the date now which we all celebrate World Wide every year as World Health Day. Currently WHO is working with more than 7000 peoples from more than 150 countries with availability of 150 countries offices, with 6 Regional Offices reporting all at the headquarter situated at GENEVA, SWITZERLAND. WHO plays a crucial role as an intermediate to coordinate with different International health agencies within the UNITED NATIONS. WHO works worldwide to promote health, keep the world safe, and serve the vulnerable. [1]
The Application or Expression of Interest which are submitted for Product Approval are assessed for the Finished Pharmaceutical Product (FFP) or Active Pharmaceutical Ingredient Master File (APIMF) is conducted at WHO Headquarters in GENEVA, SWITZERLAND. At the embryonic stage the concealing for existence of the documents are performed for FPP and APIs which ensures that the DOSSIER or the APIMF restrains the prescribed particular informations. The Applications for FPPs or APIMF are accepted by the Expert Review Panel Prequalification Team for those Manufacturer or Marketer whose documents are accomplished and attached as per the format/ checklist provided in the EOI. The applications which are found not eligible for the invitation to the Prequalification Team shows the lack of appropriate information or observance of critical deficiencies which could not be addressed by the applicant within the defined time frame.
Expert Review Panel:
The Expert Review Panel (ERP) is an self-supporting counselling body of scientific authorities, harmonized by WHO. ERP assesses the quality risks of pharmaceutical products that do not meet all stringent quality requirements, based on transparent criteria and provides advice for the purpose of aiding procurement decisions regarding time limited procurement. [2]
Products are classified in one of four risk categories:
• risk category 1 or 2: No objection to time-limited procurement[2]
• risk category 3: Objection to procurement but may be considered when there are no alternatives, [2]
• and provided the benefit outweighs the risk of procuring a product which is not fully quality assured. [2]
• risk category 4: Objection to procurement. [2]
TABLE 1
Overview of ERP Assessment Criteria: [2]
|
CONTENTS |
RISK CATEGORY 1 *No objection to procurement (risk category 1) Product described by all of the below. |
RISK CATEGORY 2 *No objection to procurement (risk category 2) Product described by any of the below |
RISK CATEGORY 3 Objection to procurement, but can be procured if benefit outweighs risk (risk category 3) Product described by any of the below |
RISK CATEGORY 4 Objection to procurement Product described by any of the below |
|---|---|---|---|---|
|
Finished product manufacture and controls |
Acceptable specifications (in-house or compendial + additional in-house tests, and verified compendial / validated in-house methods). For sterile products, manufacturing processes adequately validated |
Acceptable specifications as per official monograph but missing certain additional in-house tests; for sterile products, manufacturing processes are adequately validated |
Acceptable specification but analytical methods not sufficiently validated |
Unacceptable specification or analytical validation for a critical test parameter; for sterile products, manufacturing process not adequately validated |
|
Stability and shelf life |
The submitted data support the claimed shelf life or an acceptable shelf life during which the product will comply with acceptable specifications |
The submitted data support the claimed shelf-life or an acceptable shelf life during which the product will comply with compendial specifications |
Shelf life is supported by insufficient stability data (e.g. submission of data on only one batch of a product with potential stability problems). |
The available stability data does not allow any assignment of shelf life |
|
Safety and efficacy: For generics: Evidence of therapeutic equivalence an acceptable comparator |
Acceptable evidence of safety and efficacy OR demonstrated in vivo bioequivalence with an acceptable comparator product, OR (for oral products exempt from bioequivalence studies) acceptable multimedia dissolution data |
Bioequivalence demonstrated or for biowaiver-eligible oral products similarity in multimedia dissolution studies; the source of the comparator product is unknown or known to be outside of ICH** OR comparator itself is a generic but WHO prequalified or SRA-approved |
Bioequivalence data not submitted, but for orally administered products, multi-media dissolution data show similarity (for non-oral products other in vitro data, as applicable, indicate similarity), AND/OR comparator is a generic product not prequalified or SRA-authorized |
Efficacy and safety data not submitted, or unsatisfactory (e.g. several major deficiencies) |
|
Source and quality of active pharmaceutical ingredients (API) |
API has acceptable specifications and is manufactured at a GMP-compliant facility as inspected by WHO, SRA or PIC/s member inspectorate |
API has acceptable specifications with no major quality concern and is manufactured at a licensed site with no known GMP issues |
API has acceptable specifications but GMP issues have been identified |
API specification not acceptable for a critical test parameter such as impurities |
*applicable only to products under assessment by PQTm or an SRA (Stringent Regulatory Authority).
** ICH: The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. See: ich.org/
Documents Required for Compilation under Expression of Interest (EOI) for Product Evaluation through ERP Prequalification Team:
Basically the Procedure for submission of Application and requirement of documents for Expression of Interest are almost same for all the therapeutic category of the Product but here we are discussing about Reproductive Health Medicines.[3]
It contains 6 sections namely:
Administrative (Section 1):
Under administrative section information about Active Ingredients and the Finished Products need to be given like International Non-proprietary Names, Generic Name of the Product, Trade name or Proprietary Name if any, Dosage form of the Product, Strength per dosage unit, Route of Administration, Pharmacopoeial status of the Finished Product as well as each ingredients which are used in the manufacturing of the said finished Product, Packaging Profile of the Drug Product, Address of the Manufacturing Site of Finished Product, Details of Certificate of Suitability/ CEP Certificate of API, Regulatory Status of the Product means the License/ Permission to manufacture and sell the Product Internationally, Requirement of Certificate of Pharmaceutical Product also known as COPP, In case if the product is registered in number of countries then product approval or registration certificate of each countries need to be attached in the application, Primary and Secondary Packing of the Finished Product, Patient Information Leaflet/ Package Leaflet along with the samples of the Finished Products. [3]
Active Pharmaceutical Product (Section 2):
Details of the Manufacturer (name, physical address and country)/manufacturing site along with the GMP certificate of the Unit in which the API is manufactured, information of API status regarding the WHO Prequalification need to be given that is the API is WHO prequalified or not, Specification of the API means the Pharmacopoeial Status of the API need to be mentioned like BP/USP/EP/International Pharmacopoeia or any, API specification along with the Testing Procedure of the API from Vendor and the API tested in the Finished Product Manufacturing Site, Analytical Method Validation of the API if Pharmacopoeial then compendial and if API is Non-Pharmacopoeial in that case complete Method Validation, Certificate of Analysis of the API from Vendor and the API tested in the Finished Product Manufacturing Site, Certificate of suitability to the monograph of the European Pharmacopoeia, Open Part of Drug Master File (DMF). [3]
Finished Pharmaceutical Product (Section 3):
Finished Product Manufacturing Site GMP Status along with the details of the other GMP Inspections carried out in the last 5 Years, Finished pharmaceutical product specification with separate specifications of Release and Shelf Life, Analytical Method Specification along with the Validations of the Analytical Methods, Method of Manufacturing with Batch Size and Process Validation of the Manufacturing Process and Control, Flow diagram and brief narrative describing the manufacturing and control process of the product with relevant parameters, Stability of the Finished Product- Stability Protocol and Stability Summary Sheet with minimum 3 bathes to be provided. [3]
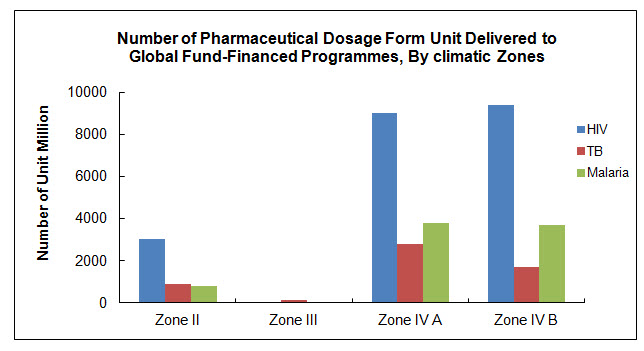
When evaluating applications WHO assumes that all the medicines prequalified will be used in all sub-zones of Climatic Zones III and IV, unless otherwise properly justified by the applicant and confirmed by WHO (see diagram below with supply data per climatic zone). Therefore, in order to safeguard product quality throughout its entire intended shelf-life, stability studies under the conditions defined for Climatic Zones IVb should be performed and the data submitted, i.e. the shelf-life should be established based on complete long-term data at 30ºC ±2ºC/75% RH ±5% RH. [4] (“Complete” refers to the length of data required at the time of dossier submission.)
Furthermore, in order to facilitate procurement decisions, the accepted storage conditions and the established shelf-life are now included in the WHO List of Prequalified Medicinal Products for each prequalified product.
For detailed guidance on stability requirements for submission of dossiers to PQTm, the PQTm quality guideline should be consulted. [4] For more general guidance on conducting stability studies, the WHO guidelines on the stability testing of finished pharmaceutical products should be consulted. [5]
The diagram below shows the number of units supplied to Zone II, Zone III, Zone IV A & Zone IV B countries for Products supplied during the Period 2007 to March 2010. [6]
Safety/Efficacy and/or Therapeutic Equivalence (Section 4):
Therapeutic equivalence (BE study), comparative dissolution profile, dissolution tests, and others, if any, to be submitted. Details of the Reference Product, In vivo bioequivalence studies conducted between the Reference and Finished Product, Result of F1 and F2 Studies, Commitment that the product used in the therapeutic equivalence study is essentially the same as the one that will be supplied (same materials from the same suppliers, same formula and same manufacturing method). [3]
Comparator products should be purchased from a well regulated market with stringent regulatory authority
A regulatory authority that is:
a) a member of ICH prior to 23 October 2015, namely: the US Food and Drug Administration, the European Commission and the Ministry of Health, Labour and Welfare of Japan also represented by the Pharmaceuticals and Medical Devices Agency; or
b) an ICH observer prior to 23 October 2015, namely: the European Free Trade Association, as represented by Swissmedic and Health Canada; or
c) a regulatory authority associated with an ICH member through a legally-binding, mutual recognition agreement prior to 23 October 2015, namely: Australia, Iceland, Liechtenstein and Norway. [7]
• Obtaining Comparator:
Comparator products should be purchased from a well regulated market with stringent regulatory authority. If the recommended comparator cannot be located for purchase from the market of one of the identified countries, the applicant should consult with WHO regarding the sourcing of an acceptable comparator product. [7]
• Information Requirements:
Within the submitted dossier, the country of origin of the comparator product should be reported together with lot number and expiry date, as well as results of pharmaceutical analysis to prove pharmaceutical equivalence. Further, in order to prove the origin of the comparator product the applicant must present all of the following documents: [7]
1. Copy of the comparator product labelling. The name of the product, name and address of the manufacturer, batch number, and expiry date should be clearly visible on the labelling. [7]
2. Copy of the invoice from the distributor or company from which the comparator product was purchased. The address of the distributor must be clearly visible on the invoice. [7]
3. Documentation verifying the method of shipment and storage conditions of the comparator product from the time of purchase to the time of study initiation[7].
4. A signed statement certifying the authenticity of the above documents and that the comparator product was purchased from the specified national market. The certification should be signed by the company executive or equivalent responsible for the application to the Prequalification Programme. [7]
• Dose Equivalence:
In case the invited product has a different dose compared to the available acceptable comparator product, it is not always necessary to carry out a bioequivalence study at the same dose level; if the active substance shows linear pharmacokinetics, extrapolation between similar doses may be applied by dose normalisation. [7]
• Fixed-dose Combination Products:
The bioequivalence of fixed-dose combination (FDC) product should be established following the same general principles. The submitted FDC product should be compared with the respective innovator FDC product as listed above. In cases where a FDC comparator product is not listed above, individual component products administered in loose combination should be used as a comparator. The principles of dose normalization as mentioned above are applicable. [7]
• Suitability of a comparator product for BCS-based biowaiver applications:
Recommendation of an API for BCS-based biowaivers is made purely on the solubility, permeability, safety and related properties of the API (Class 1 or Class 3) It does not imply that the recommended comparator product(s) will be rapidly dissolving in case of Class 1 APIs (or very rapidly dissolving in case of Class 3 API), which is a requirement for BCS based biowaiver studies. The applicant must thus ensure that the recommended comparator(s) listed on the Prequalification website is indeed suitable for a BCS based-biowaiver application before product development. [7]
Note that rapidly dissolving (or very rapidly dissolving) properties of a product are not required for in vivo bioequivalence studies. Thus, though a listed comparator product may not be suitable for BCS-based biowaiver purposes, it is still suitable for in vivobioequivalence studies[7]
• Commitment and Authorization (Section 5):
Commitment Letter, Power of attorney and Authorization for sharing information to the concern authority shall be provided. [3]
• Attachments/ Annexure (Section 6):
Annexures or Attachments shall be provided along with the Expression of Interest or Application and it shall be in PDF Format for ease to review, kindly also ensure that all the documents shall be attached with proper annexuring as marked in the application. The checklist may not be exhaustive. [3]
4. Eligibility for Prequalification (EOI) of Finished Pharmaceutical Products and Active Pharmaceutical Ingredients:
The initial step in the process of Prequalification of a Finished Pharmaceutical Product and Active Pharmaceutical Ingredient (API) is an invitation to the manufacturer to submit an Expression of Interest (EOI) for Product Evaluation. EOI is issued by the WHO according to the Therapeutic Category prescribed by the WHO Disease Programme or by the Clinical Specialist.
Presently only those Manufacturer can submit the Expression of Interest (EOI) for product evaluation who are manufacturing the below mentioned therapeutic category of the Drugs w.r.t [8] Finished Products and APIs used in the manufacturing of these Finished Product:
* Diarrhoeal Disease
* Hepatitis B and C
* HIV/AIDS
* Influenza
* Malaria
* Neglected Tropical Diseases
* Reproductive Health
* Tuberculosis [8]
5. Who Prequalification Process:
For each application for product evaluation, a manufacturer must submit a covering letter, product dossier, product sample and site master file to the WHO Prequalification Programme. Thereafter the Programme undertakes comprehensive evaluation of the quality, safety and efficacy of the product, based on the information submitted by the manufacturer, and inspection of the corresponding manufacturing site(s). Products submitted for prequalification are often multisource generics. In such cases, therapeutic equivalence with an innovator product is verified by performing a bioequivalence study. Such studies are generally carried out by an independent clinical research organization (CRO), which must therefore also be inspected and approved. [9]
The results — both positive and negative — of dossier assessments and inspections are relayed to manufacturers and CROs, together with advice, if needed, concerning corrective actions that are required if prequalification is to be achieved. [9]
WHO charges fees for applications to prequalify medicines, but with several important exemptions, including for first time applications. Generally, every product contained in an EOI is already included on the WHO Model List of Essential Medicines and/or in WHO treatment guidelines. [9]
6. Benefits For Manufacturers Of Participating In Who Medicines Prequalification [9]
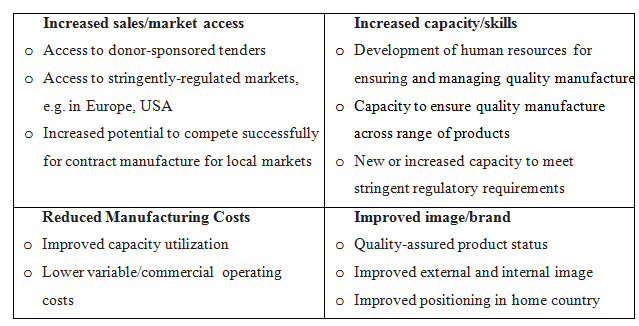
By Participating in WHO Prequalification Programme the Manufacturer gets benefit to enhance their skilfulness in developing and preparation of product dossiers and assembling manufacturing sites for inspection. As a result they will not only become better but perform good documentation for submission of Dossier for Product Evaluation in future this will also help to resolve current technical problems relating to quality-assured manufacturing of products. [9]
Table 2: Flow Chart of Prequalification Procedure [10]
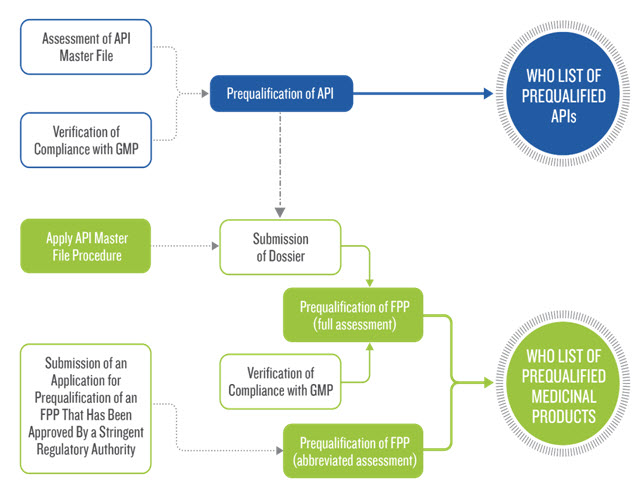
CONCLUSION : For registration of Pharmaceutical Product in WHO GENEVA the basic criteria is to compile the documents as per the requirements mentioned in the Application of Expert Review Panel (ERP) which receives the documents as per the format mentioned in the ERP and the documents attached as per the annexures. The Expert Review Panel rejects the application for registration of Pharmaceutical Product for WHO Geneva in case if the documents are not compiled properly or if any of the annexure is missed to attach along with its application, so therefore it’s mandatory to compile and submit the documents as per the format prescribed by the ERP. The Application contains the information on both the drug substance part and the drug product part as detailed above.
REFERENCES:
1. https://www.who.int/about
2. Expert Review Panel, WHO Prequalification Team: Medicines, Information Note- 08 February 2019 https://extranet.who.int/prequal/sites/default/files/documents/73_ERP_Feb2019.pdf
3. 14th Invitation to Manufacturers of Reproductive Health Medicines to Submit an Expression of Interest (EOI) for Product Evaluation by the WHO Expert Review Panel (ERP) for Reproductive Health Medicines 01 June 2018 https://extranet.who.int/prequal/sites/default/files/documents/UNFPA_14EOI_June2018.pdf
4. WHO Technical Report Series, No. 1010, 2018, Annex 10: Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. http://apps.who.int/medicinedocs/documents/s23498en/s23498en.pdf
5. WHO Technical Report Series, No. 970, 2012, Annex 4, 3.2.P.8: Guidelines on submission of documentation for a multisource (generic) finished pharmaceutical product for the WHO Prequalification of Medicines Programme: quality part
6. Requirements for Stability Studies of Finished Pharmaceutical Products, WHO Prequalification Team: Medicines, Guidance Document- 29th March 2010 https://extranet.who.int/prequal/sites/default/files/documents/27%20Stability%20requirements_March2016.pdf
7. Recommended comparator products: Reproductive Health medicines, WHO Prequalification Team: Medicines, Guidance Document 14 July 2018 https://extranet.who.int/prequal/sites/default/files/documents/Comparator-RH2018-14July.pdf
8. FPPs & APIs Eligible for Prequalification ("EOIs") https://extranet.who.int/prequal/content/products-eligible-prequalification
9. Investing In WHO Prequalification of Finished Pharmaceutical Products; Information for Manufacturer; World Health Organization. https://extranet.who.int/prequal/sites/default/files/documents/WHO_Prequalification_WHY_2.pdf
10. Flow chart for Prequalification Procedure https://extranet.who.int/prequal/content/prequalification-procedures-and-fees-0
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT editor-in-chief@pharmatutor.org
Subscribe to Pharmatutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








