

AUTHORS :
Gouranga Sundar Roy 1*, Sangita Biswas2
Department of Pharmacy, Bengal School of Technology
(A College of Pharmacy),
Sugandha, Delhi Road Near Chuchura, Hooghly - 712102,
West Bengal, India
ABSTRACT:
Scott Craniodigital syndrome is a situation that has best been found in families. The manifestations consist of uncommon head shape, boom and developmental delay, and mild webbing between the arms and toes. Less than 10 cases have been described within the literature so far. Clinical description strange dermatoglyphic patterns, boom retardation and brachycephalic have additionally been reported. Scott Craniodigital Syndrome with Mental Retardation is a very uncommon inherited disorder this is fully expressed in male best. Mental retardation (MR) is a widely used term but a contentious one. There is still a quest for appropriate terminology that doesn't companion with negative perception of affected character ad but is correct in phrases of nosology. Also debatable is the most appropriate methodology. Scott Craniodigital syndrome is believed to be inherited as an X-connected recessive genetic trait, based totally on the presence of the circumstance in males most effective. Carrier ladies have very moderate manifestation. The motion disorder related with can be managed by way of pharmacological treatment of thru physical remedy and occupational therapy to reduce disability. Some drug remedy which have been used to control.
INTRODUCTION
Scott Craniodigital disorder is a condition that has just been found in two families. The indications incorporate unordinary head shape, development and formative postponement, and mellow webbing between the fingers and toes (syndactyly) [1]. The conjunction of mental and development hindrance, strange likes, syndactyly, gentle brachycephalic and irregular palmer dermatoglyphic in every one of the 3 male kin appears to speak to a particular element [2]. Despite the fact that the disorder is dubiously reminiscent of other "head-hand" disorders, it is clinically particular from the perceived kinds of acrocephalosyndactylism: Apert's disorder, Nocks' disorder, Carpenter's disorder, Pfeiffer's disorder, Vogt's cephalodactyly and the Waardenbrug sort of acrocephalosyndactylism. Despite the fact that the youngsters detailed here externally family's cases revealed by Cozen and fume they contrast essentially in their facial appearance and level of development and mental impediment [3].
HISTORICAL NOTES
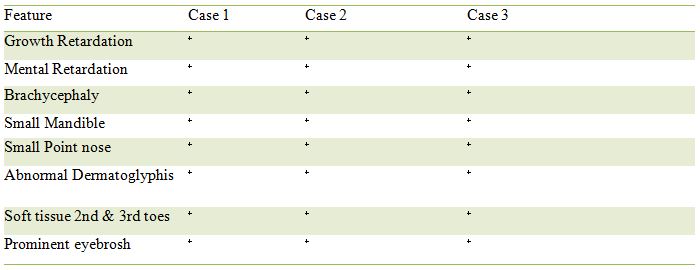
In 1971, Scott et al. portray an abnormality disorder in 3 siblings with mental impediment, development hindrance, bizarre facial appearance and syndacpilled in Table 1.Not having the option to class these side effects with one of the definitely known disorder, the creators reasoned that they comprised a different element [4]. Their perception was recorded in McKusick's list as section 31286.To our insight different cases have not been distributed from that point forward. Cutaneous syndactyly between the second and third toes of the mother of these patients drove the creators to the end X-connected latent legacy [5].
FAMILY DATA
The 3 influenced young men were the main youngsters in the sib transport [6]. The dad and mother were 46 and 31 years of age, separately at the introduction of their first youngster and were of Northern European plummet. They were not known to be connected. Their insight was typical [7]. The mother had delicate tissue syndactyly between the second and third toes reciprocally however didn't shear other element in the same way as the influenced kids [8]. She expressed that her mom had a comparable kind of syndactyly yet was uninformed of some other relative with syndactylylism [9].
EPIDEMIOLOGY
Under 10 cases have been depicted in the writing so far [10]. Clinical portrayal unusual dermatoglyphic designs, development hindrance and brachycephalic have additionally been accounted for. Scott Craniodigital Syndrome with Mental Retardation is an incredibly uncommon acquired issue that is completely communicated in male as it were. Anyway females convey a solitary duplicate of the illness quality (heterozygote's) may displays a portion of the side effects related with the confusion. The confusion has been accounted for in two skewered families in the clinical writing .Most of the indications are evident during childbirth. Under 10 cases have been depicted in the writing up until now.
ETIOLOGY
Mental impediment (MR) is a broadly utilized term however a petulant one. There is as yet a journey for fitting wording that doesn't connect with negative impression of influenced singular promotion yet is exact as far as nosology. Likewise dubious is the most suitable approach .Studies on the pervasiveness of MR have given fluctuated appraises especially for mellow MR because of ascertainment children and variable definition. Be that as it may, moderate to serious MR is evaluated at 3.8 per 1,000 and gentle MR at ~03 per 1000. Hereditary abnormalities are causative in a huge comprehension of these has expanded generously in late decades. Notwithstanding the definition, grouping and etiology of MR. There are four sorts of Scott Craniodigital disorder, for example, Craniodigital Syndrome-Mental Retardation, Scott Type, Craniodigital Syndrome of Scott, Scott Syndrome.
PATHOPHYSIOLOGY
Scott Craniodigital disorder is accepted to be acquired as a X-connected latent hereditary characteristic, in light of the nearness of the condition in guys as it were. Transporter females have extremely mellow indication .Only two families have been portrayed with the condition, so the genuine quality recurrence in obscure. Chromosomes, which are available in the core in the human cells, convey the hereditary data for every person. Human body cells ordinarily have 46 chromosomes. Pair of human chromosome is numbered from 1 through 22 and the sex chromosomes assigned X and Y. Guys have one X and one Y chromosomes and Female have two X chromosome. Every chromosome has a short arm assigned "P" and a long arm assigned Chromosome are further sub-separated into numerous groups that are numbered. For the model, "chromosome 11p13" alludes to bond 13 on the short arm of chromosome 11 .The numbered groups indicate the area of the thousand quality that are available on every chromosome. Hereditary infections are controlled by the mix of quality for a specific base pair that are on the chromosome gotten the dad and the mother. Latent hereditary issue happen when an individual acquires the equivalent strange quality for a similar characteristic from each parent [20]. On the off chance that an individual gets one ordinary quality and one quality for the sickness, the individual will be a bearer for the infection, however as a rule won't show manifestations. The hazard for two bearer guardians to both pass the deficient quality and thusly, have an influenced kid is 25% with every pregnancy the possibility for a youngster to get typical qualities from the two guardians and be hereditarily typical for that specific attribute is 25%. The hazard is the equivalent for guys and females. All people convey 4-5 irregular qualities. Guardians who are close family members (consanguineous) have a higher possibility then inconsequential guardians to both convey the equivalent unusual quality.
SYMPTOMS Hereditarily :
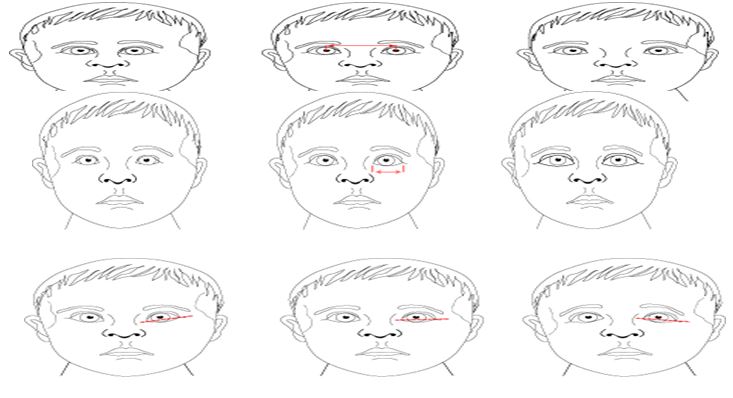
That expansion the hazard to have kids h a passive hereditary disorderd.X-connected latent hereditary issue are condition brought about by an unusual hereditary X-chromosome .Female have two X chromosome however one of the X chromosome is "killed" and the entirety of the qualities on that chromosome are inactivated. Females who have an illness quality present on one of their X chromosome are bearers for those clutters. Transporter families as a rule don't show manifestations of the scatters since it is generally the X chromosome with the strange quality that is killed. A male has one X chromosome and the acquires a X chromosome that contains an infection quality, he will build up the illness quality, will build up the malady. Guys with X connected turmoil pass the malady quality to the entirety of their little girls, who will be transporters. A male can't pass a X-connected quality to his child s since guys consistently pass their Y chromosome instated of their X chromosome to male posterity. Females bearers of X-connected disarranges have a 25% opportunity to have transporters drafts such as themselves, a 25% opportunity to have a child influenced with the illness and a 25% opportunity to an unaffected child. In female who acquire a solitary duplicate of the illness quality for Scott Craniodigital disorder (Heterozygote's),disease characteristics on the X chromosome may not generally be covered by the ordinary quality on the typical quality on the other X chromosome ; therefore , these females may shows a portion of the side effect related with this issue . Heterozygous is condition in which an individual has two contrast qualities (alleles) at a similar spot on coordinated chromosome. An person that is heterozygous for a specific attribute from one parent. An individual heterozygous for an innate issue delivered by a passive quality won't show the malady or won't show the infection or will have milder from. Dysmorphic face :- Face pelvic scapula dysplasia short stature heart deformity Craniofacial oddities short stature hyperkalemia acidosis short status locking fingers short status mental impediment eye inconsistencies short mental hindrance eye abandons short stature . Orbital Placement: Hypertelorism is defined by an increase interpupillary distance. Hypertelorism (right); normal (middle); hypotelorism (left) .Palpebral Fissure Length:-Often this length is actually measures and plotted.Short (left); normal (middle); large (right).Palpebral Fissure Slant:-This varies greatly with ethnic origin. Up (left); normal (middle); down (right).Epicanthal folds:-Many variations exist. The boy on the left does not have folds. On the right image, the effect of the epicanthal fold extending above the inner can thus is illustrated.
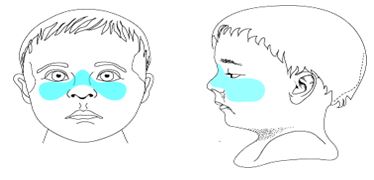
Mid-face Hypoplasias:-Midface hypoplasia (blue region below) is essentially the same thing as maxillary bone region hypoplasia. Maxillary retrusion is another way of thinking about mid-face hypoplasia. Depressed nasal bridge (not illustrated here) is often another component of mid-face hypoplasia .
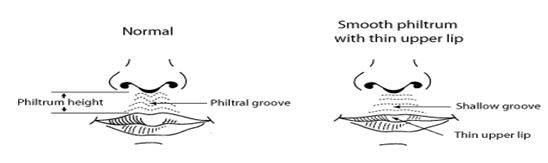
Philtrum shape The philtral groove can be through of as representing the resultant folding effects of bilateral, inwardingly migrating neural crest cells that met at the midline. Smooth patterns however can also be normal variants or can be associated with genetic syndrome or caused by teratogens such as alcohol.
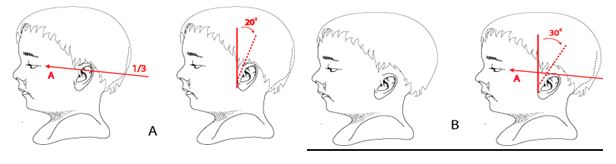
Ear Placement Images a show normal ear position. Low set ear are positioned below the horizontal lines as illustrated in B. Low-set ears are often posteriorly rotated reflecting in the normal anterior rotation that occurred during ear development.
GAIT ATAXIA : Gait ataxia Gait Ataxia gait disorder, Neurologic find out more search to find out more about Scott Craniodigital Syndrome [34]. GAIT APRAXIA: Gait ataxia Gait Ataxia gait disorder, Neurologic find out more search to find out more about Scott Craniodigital Syndrome .
APRAXIA : Apraxia in a bilateral frontal lobe disorders. It is characterised by an inability to initiate the process of walking, despite the power and coordination of the legs being normal when tested in the seated or lying position. The gait is broad-based with short steps with a tendency to fall backwards. It was originally described in patients with frontal lobe tumors, but is now more commonly seen in patients with cerebrovascular disease.
DIAGNOSIS
The diagnosis of Scott Craniodigital Syndrome may be made at birth or during early infancy based upon a thorough clinical evaluation and characteristic physical finding. Specialized testing, such as certain advance imaging techniques, may also be conducted to detect or characterize particular findings that may be associated with the disorder .
DERMATOGLYPHICS:
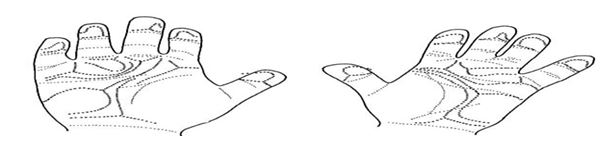
The dermatoglyphic pattern in each child was unusual. The most striking abnormality was the distal position of the palmer axial triradius associated with a large hypothenar pattern. Each boy had an atd angle greater than 70 degree on at least one hand. The greater then 2 stander deviation from the mean of an age and sex-match control group for the 2 younger boys. In addition to the digital syndactyly[11] , each boy had an abnormal creasing pattern on some fingers and unusual minor features . The major dermatoglyphic pattern is outline in Fig. The dermatoglyphic pattern of the father was considered normal . The mother , however , had small ulnar loops and one arch on her fingertips with an unusually low total ridge count of 32 , A dermatoglyphic tendency was noted on each hand [12].
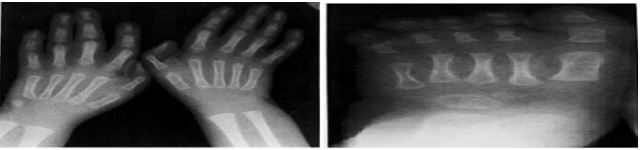
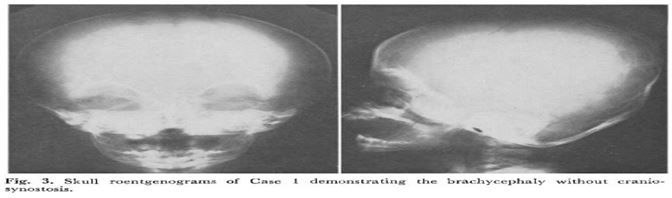
RADIOLOGIC FINDING : Roentgen graphic studies demonstrated identical abnormalities in each child . There was moderated to severbrachycephaly without evidence of premature fusion of cranial sutures . This was associated in each case with roentgenogramphic hypertelorism , an unusual number of Wormian bones increased calvarial vascular pattern and a borderline shorted mandible [13] . Skeletal maturation was below average but within normal limits for each child. Each patient had mild hind foot varus deformity and L5-S1 spina bifida osculates [14].
TREATMENT COUNSILING OF MENTAL RETARDATION PATIENT:-
Recently pediatricians have become more interested in the retarded child and his family and have come to realize that they have been ill-prepared to face the many problems that continually arise. A basic understanding of the role of parents and their aspirations, coupled with knowledge of interviewing, techniques will enable them to face these problems with greater assurance. In addition, knowledge of the community facilities and resources is necessary [15]. This will entail direct contact with schools, parent groups, and state hospitals for the retarded as well as state and local agencies which deal with the retarded. Individual patient’s condition, utilizing many assessment tools:
PSYCHOTHERAPY:
Psychotherapy deals successfully with the emotional problems and problems of maladjustment, as well as psychological symptoms. It is a well-established fact that mentally subnormal people demonstrate a number of psychological problems and complexes which can be reduced by psychotherapy alone. Under these circumstances, psychotherapy becomes a very effective method of treatment. Usually, individual psychotherapy, group psychotherapy, behavior modification and observational learning are included under psychotherapy.
MEDICINE REUQIRMENT: - The movement disorder associated with can be managed by pharmacological treatment of through physical therapy and occupational therapy to reduce disability. Some drug treatment that have been used to control include : (5HTP) , Idebenone , Amantadine ,, Physotigmine , L-carnitine orderivatives,Trimethoprim / Sulfamethoxazole , Busprine and a combination of Coenzye Q10 and vitamin E.
LABORATORY studies : The following blood determination revalued normal values: red cell mass, white cell morphology, platelet count, calcium, phosphorus, urea, alkaline phosphatase and amino acid. Routine urinalysis was manner with Benedict’s reagent, 2, 4- dinitrophenylhydrazine and sodium-cyanide sodium-nitroprusside reagent. The excretion pattern of phenolic acids, indole , porphyrins and amino acids were normal by mucopolysaccharides values were normal. Cerebral spinal fluid gave normal quantitative values for glucose, protein, chloride and amino acids [16].
CASE REPORT
S.H was delivered after 36 weeks of uneventful gestation by cesarean section after premature rupture of the fetal membranes . He was the first child ( birth wt.1990 g , length 46 cm) . Initial as phyxia was quickly alleviated by intubation and CPAP. Hyperbilirubinemia (maximum 388µmol/liter) necessitated an exchange transfusion on the first day of the life . Anomalies of the face, hand and feet were noted at birth . Chromosomes were normal . Consanguinity was denied by the parents . Family history is unremarkable [17].

At 12 months , the patient was referred for mental retardation , sbrachycephaly with micrognathia and telecanthus (fig 1) . Length was 77cm (3rd to10th centile). In addition he had syndactylies of the fingers and toes 2-4 .There were also extensive dermal ridge anomalies with bilateral simian crease . The other anomalies are compared to those of the patient of Scott et al [1971] In Table1 . Delayed skeletal maturation made us first assume the presence of hypothyroidism . The first TRH-test showed values consistent with latent hypothyroidism and treatment with thyroid hormone (10µg T3 and 40µg T4) was instituted [18].
CONCLUSION
The pattern of anomalies in our patient resemble that describe by Scott et al [1971]. The only additional manifesting is a considerably delayed maturation of the skeleton. Hypothyroidism, as a possible cause of this was ruled out. Similarly severe delay of ossification was observed by Eiken et al [1984] in 3 brothers with dwarfism and abnormal modeling the bones of hands and feet. However, there was no mental retardation and no syndactyly in these patients. The diagnosis which led to the referral of the patient describe above to our institute was oculo-dento-osteo dysplasia (ODOD). Indeed, it must be admitted that the facial anomalies suggest the diagnosis of ODOD the syndactyly requires this with broadening of the cortical substance was absent in our patient. Moreover, patients with ODOD the syndactyly is always found between the fourth and fifth toes .The mother of our patients had mild syndactyly between toes 2 and 3, precisely as in the case described by Scoott et al.[1971] . We therefore concur with Scott et al [1971] that this entity may be an X-inked recessive trait.
REFERENCE
1. Lorenz P, Hinkel GK, Hoffmann C, Rupprecht E. The craniodigital syndrome of Scott: report of a second family. Am J Med Genet. 1990; 37:224-226.
2. Scott CR, Bryant JI, Graham BC. A new craniodigital syndrome with mental retardation. J Pediatr. 1971; 78:658-663.
3. Shevell M, Ashwall S, Donley D, et al. Practice parameter: evaluation of the child with global developmental delay. Neurology. 2003; 60:367-380.
4. Soekarman D, Volcke P, Fryns JP. On the nosology of the craniodigital syndromes: report of a family and review of the literature. Genet Couns. 1997; 8:217-222.
5. Toriello HV, Higgins JV. Craniodigital syndromes: report of a child with Filippi syndrome and discussion of differential diagnosis. Am J Med Genet. 1995; 55:200-204.
6. Ali MS, Mukherjee S, Roy D, Pal G, Makar S. Asparagus Racemosus, a Climbing Ayurvedic Medicinal Plant: Review on its Cultivation, Morphology and Medicinal Significance. PharmaTutor. 2018 Dec 1;6(12):46-54.
7. Nag M, Mukherjee S, Roy D, Paul RK, Sahidul M. Extraction and Chemical tests on Cicer Arietinum seed collected from North Bengal Region of West Bengal, India.
8. Mir SA, Mukherjee S, Makar S, Pal G. Cucurbitacins a Vibrant Triterpenoid: A Review on its Anticancer Property. PharmaTutor. 2019 Feb 1;7(2):43-54.
9. Mukherjee S, Roy D, Das S. Apicoplast: a brilliant focus for antimalarial drug development. PharmaTutor. 2018 May 1;6(5):13-22.
10. Pal D, Ahuja C, Mukherjee S. Celsia coromandeliane Vahl-A New Biomarker with Immense Pharmaceutical Applications. Journal of Drug Delivery and Therapeutics. 2019 Jun 29;9(3-s):1109-15.
11. Chitayat D, Meunier CM, Hodgkinson KA, Azouz ME (1991): New syndrome? Robin sequence with facial and digital anomalies in two half-brothers by the same mother. Am J Med Genet 40:167-172.
12.Filippi G (1985): Unusual facial appearance, microcephaly, growth and mental retardation, and syndactyly. A new syndrome? Am J Med Genet 22:821-824.
13. Kelly TE, Kurson L, Wyatt J (1993): Microcephaly and digital anomalies: A newly recognized syndrome of recessively inherited mental retardation. Am J Med Genet 45353-355.
14. Lorenz P, Hinkel GK, Hoffman D, Rupprecht E (1990): The craniodigital syndrome of Scott: Report of a second family. Am J Med Genet 37:224-226.
15. Meinecke P (1993): Short stature, microcephaly, characteristic face, syndactyly, and mental retardation: The Filippi syndrome. Genet Couns 4:147-151.
16. Scott CR, Bryant JI, Graham CB (1971): A new craniodigital syndrome with mental retardation. J Pediatr 78:658-663.
17. Woods CG, Crouchman M, Huson SM (1992): Three sibs with phalangeal anomalies, microcephaly severe mental retardation, and neurologic abnormalities. J Med Genet 29:500-502.
18. Zerres K, Rietschel M, Rietschel E, Majewski F, Meinecke P (1992): Postnatal short stature, microcephaly, severe syndactyly of hand and feet, dysmorphic face, and mental retardation: A new syndrome?: J Med Genet 29:269-271.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT admin@pharmatutor.org
FIND OUT MORE ARTICLES AT OUR DATABASE








