
 About Authors:
About Authors:
ANUJ SINGH
VINAYAKA MISSION’S UNIVERSITY
Salem – 636308, Tamil Nadu
anuj.dra@gmail.com
ABSTRACT:-
Healthcare Regulatory Affair Professional with their experience and strong motivation to excel in the Regulatory field has an ability to motivate & give support and strengthen to teams members, involved in process of product registration. Professional has taught a self-starter with the proven ability to prioritize and manage projects in a busy, fast-paced, multitasking environment, along with their experience in establishing relationship with the decision makers & coordinator. The Pharmaceutical industries are among the most highly regulated industries in the country. As India is growing very rapidly in pharmaceutical sector, there is a need of regulatory affairs professionals to cater the current needs of industries for the global competition. Regulatory affairs professionals are the link between pharmaceutical industries and worldwide regulatory agencies. They are required to be well versed in the laws, regulations, guidelines and guidance of the regulatory agencies. There is a growing need to incorporate the current requirements of pharmaceutical industries in the standard curriculum of pharmacy colleges to prepare the students with the latest developments to serve the industries. The present article discusses the regulatory education and its need, learning resources, courses available, syllabus contents and job opportunities in regulatory affairs.
[adsense:336x280:8701650588]
Reference Id: PHARMATUTOR-ART-1405
Guideline for Submitting Documentation for Packaging for Human Drugs and Biologics."
Toxicological data on these materials would be included under this type of DMF, if not otherwise available by cross reference to another document.
IV. C.1.d.Type IV Excipient, Colorant, Flavor, Essence, or Material Used in Their Preparation
Each additive should be identified and characterized by its method of manufacture, release specifications, and testing methods.
Toxicological data on these materials would be included under this type of DMF, if not otherwise available by cross reference to another document.
Usually, the official compendia and FDA regulations for color additives (21 CFR Parts 70 through 82), direct food additives (21 CFR Parts 170 through 173), indirect food additives (21 CFR Parts 174 through 178), and food substances (21 CFR Parts 181 through 186) may be used as sources for release tests, specifications, and safety. Guidelines suggested for a Type II DMF may be helpful for preparing a Type IV DMF. The DMF should include any other supporting information and data that are not available by cross reference to another document.
IV. C.1.e.Type V: FDA Accepted Reference Information
FDA discourages the use of Type V DMF's for miscellaneous information, duplicate information, or information that should be included in one of the other types of DMF's. If any holder wishes to submit information and supporting data in a DMF that is not covered by Types I through IV, a holder must first submit a letter of intent to the Drug Master File Staff (for address, see D.5.a. of this section). FDA will then contact the holder to discuss the proposed submission.
IV. C.2. General Information and Suggestions
IV. C.2.a. Environmental Assessment
Type II, Type III, and Type IV DMF's should contain a commitment by the firm that its facilities will be operated in compliance with applicable environmental laws. If a completed environmental assessment is needed, see 21 CFR Part 25.
IV. C.2.b. Stability
Stability study design, data, interpretation, and other information should be submitted, when applicable, as outlined in the "Guideline for Submitting Documentation for the Stability of Human Drugs and Biologics."
IV. D. Format, Assembly, and Delivery
IV. D.1.
An original and duplicate are to be submitted for all DMF submissions.
Drug Master File holders and their agents/representatives should retain a complete reference copy that is identical to, and maintained in the same chronological order as, their submissions to FDA.
IV. D.2.
The original and duplicate copies must be collated, fully assembled, and individually jacketed.
Each volume of a DMF should, in general, be no more than 2 inches thick. For multivolume submissions, number each volume. For example, for a 3 volume submission, the volumes would be numbered 1 of 3, 2 of 3, and 3 of 3.
IV. D.3.
U.S. standard paper size (8-1/2 by 11 inches) is preferred.
Paper length should not be less than 10 inches nor more than 12 inches. However, it may occasionally be necessary to use individual pages larger than standard paper size to present a floor plan, synthesis diagram, batch formula, or manufacturing instructions. Those pages should be folded and mounted to allow the page to be opened for review without disassembling the jacket and refolded without damage when the volume is shelved.
IV.D.4.
The agency's system for filing DMF's provides for assembly on the left side of the page. The left margin should be at least three fourths of an inch to assure that text is not obscured in the fastened area. The right margin should be at least one half of an inch. The submitter should punch holes 8 1/2 inches apart in each page. See the page measurements shown in the following figure:
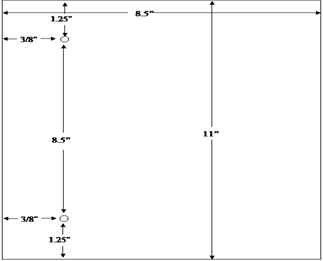
IV.D.5. Delivery to FDA
IV.D.5.a.
Drug Master File submissions and correspondence should be addressed as follows:
Drug Master File Staff
Food and Drug Administration
5901-B Ammendale Rd.
Beltsville, MD 20705-1266
IV.D.5.b. Delivery charges to the above address must be prepaid.
V. AUTHORIZATION TO REFER TO A DRUG MASTER FILE
V. A. Letter of Authorization to FDA
Before FDA can review DMF information in support of an application, the DMF holder must submit in duplicate to the DMF a letter of authorization permitting FDA to reference the DMF. If the holder cross references its own DMF, the holder should supply in a letter of authorization the information designated by items 3, 5, 6, 7, and 8 of this section. The holder does not need to send a transmittal letter with its letter of authorization.
The letter of authorization should include the following:
- The date.
- Name of DMF holder.
- DMF number.
- Name of person(s) authorized to incorporate information in the DMF by reference.
- Specific product(s) covered by the DMF.
- Submission date(s) of 5, above.
- Section numbers and/or page numbers to be referenced.
- Statement of commitment that the DMF is current and that the DMF holder will comply with the statements made in it.
- Signature of authorizing official.
- Typed name and title of official authorizing reference to the DMF.
V. B. Copy to Applicant, Sponsor, or Other Holder
The holder should also send a copy of the letter of authorization to the affected applicant, sponsor, or other holder who is authorized to incorporate by reference the specific information contained in the DMF. The applicant, sponsor, or other holder referencing a DMF is required to include a copy of the DMF holder's letter of authorization in the application.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
VI. PROCESSING AND REVIEWING POLICIES
VI. A. Policies Related to Processing Drug Master Files
VI. A.1.
Public availability of the information and data in a DMF is determined under 21 CFR Part 20, 21 CFR 314.420(e), and 21 CFR 314.430.
VI. A.2.
An original DMF submission will be examined on receipt to determine whether it meets minimum requirements for format and content. If the submission is administratively acceptable, FDA will acknowledge its receipt and assign it a DMF number.
If the submission is administratively incomplete or inadequate, it will be returned to the submitter with a letter of explanation from the Drug Master File Staff, and it will not be assigned a DMF number.
VI. B. Drug Master File Review
A DMF IS NEVER APPROVED OR DISAPPROVED.
The agency will review information in a DMF only when an IND sponsor, an applicant for an NDA, ANDA, or Export Application, or another DMF holder incorporates material in the DMF by reference. As noted, the incorporation by reference must be accompanied by a copy of the DMF holder's letter of authorization.
If FDA reviewers find deficiencies in the information provided in a DMF, a letter describing the deficiencies is sent to the DMF holder. At the same time, FDA will notify the person who relies on the information in the deficient DMF that additional information is needed in the supporting DMF. The general subject of the deficiency is identified, but details of the deficiency are disclosed only to the DMF holder. When the holder submits the requested information to the DMF in response to the agency's deficiency letter, the holder should also send a copy of the accompanying transmittal letter to the affected persons relying on the DMF and to the FDA reviewing division that identified the deficiencies. The transmittal letter will provide notice that the deficiencies have been addressed.
VII. HOLDER OBLIGATIONS
Any change or addition, including a change in authorization related to specific customers, should be submitted in duplicate and adequately cross referenced to previous submission(s). The reference should include the date(s), volume(s), section(s), and/or page number(s) affected.
VII. A. Notice Required for Changes to a Drug Master File
A holder must notify each affected applicant or sponsor who has referenced its DMF of any pertinent change in the DMF (21 CFR 314. 420(c)). Notice should be provided well before making the change in order to permit the sponsor/applicant to supplement or amend any affected application(s) as needed.
VII. B. Listing of Persons Authorized To Refer to a Drug Master File
VII. B.1.
A DMF is required to contain a complete list of persons authorized to incorporate information in the DMF by reference [21 CFR 314.420(d)]. The holder should update the list in the annual update. The updated list should contain the holder's name, DMF number, and the date of the update. The update should identify by name (or code) the information that each person is authorized to incorporate and give the location of that information by date, volume, and page number.
VII. B.2.
Any person whose authorization has been withdrawn during the previous year should be identified under a suitable caption.
VII. B.3.
If the list is unchanged on the anniversary date, the DMF holder should also submit a statement that the list is current.
VII. C. Annual Update
The holder should provide an annual report on the anniversary date of the original submission. This report should contain the required list as described in B.1., and should also identify all changes and additional information incorporated into the DMF since the previous annual report on the subject matter of the DMF. If the subject matter of the DMF is unchanged, the DMF holder should provide a statement that the subject matter of the DMF is current.
Failure to update or to assure FDA annually that previously submitted material and lists in the DMF remain current can cause delays in FDA review of a pending IND, NDA, ANDA, Export Application, or any amendment or supplement to such application; and FDA can initiate procedures for closure of the DMF (see Section IX).
VII. D. Appointment of an Agent
When an agent is appointed, the holder should submit a signed letter of appointment to the DMF giving the agent's name, address, and scope of responsibility (administrative and/or scientific). Domestic DMF holders do not need to appoint an agent or representative, although foreign DMF holders are encouraged to engage a U.S. agent.
VII. E. Transfer of Ownership
To transfer ownership of a DMF to another party, the holder should so notify FDA and authorized persons in writing. The letter should include the following:
- Name of transferee
- Address of transferee
- Name of responsible official of transferee
- Effective date of transfer
- Signature of the transferring official
- Typewritten name and title of the transferring official.
The new holder should submit a letter of acceptance of the transfer and an update of the information contained in the DMF, where appropriate. Any change relating to the new ownership (e.g., plant location and methods) should be included.
VIII. MAJOR REORGANIZATIONOF A DRUG MASTER FILE
A holder who plans a major reorganization of a DMF is encouraged to submit a detailed plan of the proposed changes and request its review by the Drug Master File Staff. The staff should be given sufficient time to comment and provide suggestions before a major reorganization is undertaken.
IX. CLOSURE OF A DRUG MASTER FILE
A holder who wishes to close a DMF should submit a request to the Drug Master File Staff stating the reason for the closure. See Section IV.D.5.a for the address.
The request should include a statement that the holder's obligations as detailed in Section VII have been fulfilled.
The Agency may close a DMF that does not contain an annual update of persons authorized to incorporate information in the DMF by reference and a list of changes made since the previous annual report. The holder will be notified of FDA's intent to close the DMF.
Many of the guidelines referred to in the text and a current list of available guidelines may be obtained from the following.
Basic Details of Stability Studies Concept:
Stability Studies:
In our universe, no example of stability can be found; everything changes, every thing is transfer, everything evolves. We are thus living in permanent state of instability.
When we deal with the drugs and their stability, we must investigate the origin of their instability and its effects on their toxicological and therapeutic activity.
Kinetics of transformations of various molecules physical, chemical or even technological transformations.
When a drug is being manufactured, the various components have a certain internal energy and certain reactivity. It is thus possible to be define an entropy for each component, this entropy being known only to within a constant factor.
This system is not isolated; it is subjected to various possible actions from the environment.
* Physical: Temperature, pressure, humidity, radiation.
* Chemical: Action of oxygen, action of water, acids, bases
Under these various energy-carrying influences, drugs will alter more or less rapidly, which may modify their toxicology-pharmacological activity.
STABILITY STUDIES
Helps in generate information, which permits well-considered proposals to be made for the shelf life of drug substance and products, and recommended storage conditions.
Stability data are required to be submitted as a part of the dossier submitted to the regulatory agencies for licensing approval. Hence it is an unavoidable activity in the drug industry.
The application of the science of degradation kinetics of active ingredients in the medicines was initiated in 1950’s. From these studies it was realized that most drug substances were inherently unstable molecules.
Around this period, the manufacturers started developing formulations in wide range of packaging materials and started selling their products to an increasing number of countries. While doing so, it was felt that there was need for conducting standardized stability studies before marketing to assure that optimally stable molecules were manufactured, distributed and supplied to the patient.
To assist in execution of stability testing, the authorities in some countries obliged the manufacturers by drawing up stability testing guidelines. These guidelines were mainly issued in 1980’s.
The guidelines, while addressing basic issues, spelt out the stability data requirements for application dossier and broadly outline the steps for execution.
However, these individual country guidelines varied in concepts, requirements and point of emphasis which subsequently proved to be a bottleneck in the drive by various manufacturers to market and register their products in more than one country- a direct fallout of globalization.
Towards the end of 1980’s the process of the harmonization of regulatory requirements begun through bilateral meetings between the officials and experts from US, Japan and EC. This culminated in holding of a symposium under the head – “Stability testing, new trends and requirements” June 5-7darmstadt, Germany. A preliminary draft was prepared at this meeting, which was discussed on 4th November 1993 at Brussels by an Expert working group and reported to Quality workshop of the first international conference on Harmonization held at the same place from 5-7 November 1991. Subsequently the guidelines “Stability Testing of New Drug Substances and Products” finalized on 27th October 1993 in Orlando.
WHO in the meantime, being an observer to the ICH, felt that the ICH parent stability guideline was unfit for universal application. The reasons were
1. The guideline did not address the extreme climatic conditions found in many countries.
2. It only covered new drug substances and products and not the already established products that were in circulation in the WHO umbrella countries.
They come up with the separate “Guideline for Stability Testing of Pharmaceutical Products containing well Established Drug Substances in Conventional Dosage Forms”.
USFDA on its part has published the guidance document entitled “Expiration Dating and Stability Testing of Solid Oral Dosage Form Drugs Containing Iron” in June 1997. in 1998, it has issued a draft version of the guidance for industry under the title “Stability Testing of Drug Substances and Drug Products”. The guidance discusses stability for NDA, ANDA and IND.
SOME OF THE INDIVIDUAL COUNTRY GUIDELINES ISSUED IN 1980’s
|
COUNTRY |
Name of the Guidelines |
Year of Introduction |
|
Japan |
Standards for stability testing of new drugs |
1980-1984 (Rev) |
|
UK |
Guidance notes on applications for product licenses (HMSO) |
1984 |
|
Zimbabwe |
Guidelines for the stability testing of drugs |
1985 |
|
USFDA |
Submitting documentation for the stability of Human Drugs and Biological |
1987 |
|
Ethiopia |
Requirements of the registration of drug for human use |
1986 |
|
EC |
Stability testing on active ingredients and finished products |
1988 |
Test Conditions:
ICH guidelines take the accelerated test condition to be 15° C above the long term testing temperature. GMP WHO the difference in accelerated and long term storage is only 10° C.
GLOBAL CLIMATE ZONES
|
ZONE |
ZONE I Moderate |
ZONEII Mediterranean |
Zone III Hot/Dry |
Zone IV Very hot/Moist |
|
Kinetic average temperature |
21° C |
25° C |
30° C |
30° C |
|
Yearly average relative humidity |
45% RH |
60% RH |
35% RH |
70% RH |
DISTRIBUTION OF WORLD NATIONS INTO DIFFERENT ZONES
|
Region |
Zone I and II Countries |
Zone III and IV Countries |
|
America |
Argentina, Bolivia, Chile, Canada, Mexico, Peru, Uruguay and USA |
Barbados, Brazil, Costa Rica, Dominican Republic, Ecuador, Jamaica, Columia, Cuba, Panama, Paraguay, Puerto Rico, Venezuela. |
|
Asia |
Afghanistan, Armenia, Azerbaijan, China, Georgia, Iran, Israel, Japan, Kazakhstan, Korea, Lebanon, Nepal, Syria, Turkey, Uzbekistan |
Hong Kong, India, Bangladesh, Iraq, Jordan, Qatar, Kuwait, Malaysia, Maldives, Myanmar, Saudi Arabia, Singapore, Srilanka, Taiwan, Thailand, UAE, Vietnam, and Yemen. |
|
Africa |
Egypt, Algeria, Tunisia, Libya, Morocco, Namibia, Rwanda, South Africa, Zambia, Zimbabwe. |
Angola, Ghana, Cameroon, Kenya, Liberia, Niger, Senegal, Central African Republic. |
|
Australian / Oceanic |
Australia, New Zealand |
Fiji, Society Islands, Marshould Islands, Samoa, Tonga |
Recommended Stability Storage conditions for various Products in Zone I and II
Stability requirements for marketing applications / Regulatory submission;
Data from formal stability studies are to be provided on at least three batched of the substance. The batches manufactured to a minimum of scale should be by the same synthetic route and use a method of manufacture and procedure and packing that simulates the final process to be used on a production scale.
Batches will be needed to be selected to comply with current FDA site specific stability requirements. To eliminate the need to provide additional data on the first three production scale batches manufactured at the commercial site, primary stability batches would need to be made at production scale at the proposed site of commercial manufacture.
STORAGE CONDITIONS
|
Storage Condition |
Tolerance |
Relation for inclusion |
|
-20° C |
± 5° C |
Freezer |
|
5° C/Ambient RH |
± 3° C |
Refrigerated Storage condition |
|
25° C/60% RH |
± 2° C/ ± 5% RH
|
(ICH) Long Term Storage Condition |
|
30° C/60% RH |
± 2° C/ ± 5% RH
|
Long Term/ Intermediate Condition |
|
40° C/75% RH |
± 2° C/ ± 5% RH |
Accelerated Testing Condition |
MANAGING STUDIES
Incubator Management:
Environmental chambers used for the storage of stability samples should be validated for the purpose. A monitoring system should be in place which can provide a record of the temperature and humidity measurement with in the chamber.
Sampling:
Samples to be supplied for analysis at each scheduled test point on a study should be withdrawn from storage randomly. The requisite number of finished product primary containers for testing at each time point should be drawn from storage. These are calculated based on the testing to be performed and the details of samples drawn should be recorded in study records. Where additional samples are required the reason for this should be documented.
Pre-test Storage:
This refers to the condition at which samples are stored after removal from the incubator and prior to commencement of testing. It is recommended that whilst samples await test the standard pre-test storage condition should be controlled room temperature. There is no requirement to store samples protected from light unless the product is known to require light protection. Products that require a lower (or frozen) pre-test storage, should be assessed and the pre-test storage documented on a case by case basis.
Transportation:
Samples requiring transport prior or testing should wherever possible be maintained at their designated pre-test storage condition during transport. Where this is not feasible, documented evidence of the range of conditions likely to be experienced by the samples in transit should exist. Additionally, maximum allowable transit times and an acceptable range of conditions should be established before samples are transported. When transported samples exceed any of the established transport parameters their continued validity as representative logging device should be included with the sample in transit.
For samples which are normally stored frozen, transit temperatures should ensure that there is no change of state in the samples. To establish whether or not thawed in transit a visual inspection is required immediately after transport of the samples, and if they have thawed, their continued validity as representative samples needs to be documented.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Testing:
Atypical and Out of Specification Results:
All atypical results and all OOS results on samples stored within recommended storage conditions and shelf life must be investigated, evaluated and clearly documented, according to procedure. If an OOS result, obtained on samples stored under accelerated conditions or beyond shelf life, is expected (e.g. a low assay follows the trend established for other samples and conditions) no investigation is required, but the documentation must include a statement to acknowledge that the result has been assessed, is valid and acceptance.
Reporting Stability Results:
The results obtained from stability testing should be assessed and reported, including any decisions made. This should include an assessment of any trends or outlines observed, as well as a comparison against the end of life specification.
Reference should be made to the predicted effects of the product change(s) on the product stability, and to whether or not these effects have been seen in practice.
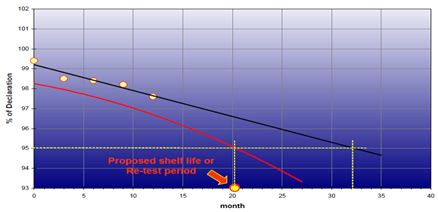
Notification of Stability Failures:
Failure in studies running in support of Clinical Trials should be notified within 24 hrs to the person responsible for the CTS The failure of post approval stability studies to meet specification either at end of life, or during storage, should be notified to FDA. For studies run in support of NDAs, the FDA should be notified using the Field Alert mechanism. In European markets the relevant licensing authority should be advised of the failure. The QA and manufacturing area should be contacted also to notify them of the failure within 24 hrs and to participate in the investigation and corrective action. All details should be documented in the relevant project of study file.
Testing scope for DRUG SUBSTANCES
• Physical-chemical properties
– Appearance
– Water content
– pH
– Color / clarity of solution
– Thermo analytical stability
» Melting point
» Polymorphism
• Chemical properties
– Assay
– Degradation products
• Microbial properties
– (Microbial purity)
Testing scope for TABLETS
• Physical-chemical properties
– Appearance
– Mean mass
– Water content
– Hardness
– Disintegration
– Dissolution
• Chemical properties
– Assay
– Degradation products
• Microbial properties
– Microbial purity
• Container closure system properties
– Functionality tests (e.g. extraction from blister)
Testing scope for CAPSULES
• Physical-chemical properties
– Elasticity
– Mean mass
– Mean filling mass
– Water content (Capsule and content)
– Disintegration
– Dissolution
• Chemical properties
– Assay
– Degradation products
• Microbial properties
– Microbial purity
• Container closure system properties
– Functionality tests (e.g. extraction from blister)
Testing scope for oral LIQUID FORMS
• Physical-chemical properties
– pH
– Color & clarity of solution
– Loss on weight
– Viscosity
– Particle size distribution (for oral suspensions only)
• Chemical properties
– Assay
– Degradation products
– Content preservatives
– Degradation preservatives
– Content antioxidants
• Microbial properties
– Microbial purity
• Container closure system properties
– Functionality tests
Testing scope for LIQUID FORMS for inj. and PARENTERALIA
• Physical-chemical properties
– pH
– Loss on weight
– Color & clarity of solution
• Chemical properties
– Assay
– Degradation products
– Content preservatives
– Degradation preservatives
– Content antioxidants
• Microbial properties
– Microbial purity
• Container closure system properties
– Functionality tests
Testing scope for SEMI LIQUID FORMS
• Physical-chemical properties
– Appearance, odor, homogeneity, consistency
– Loss on weight
– Viscosity
– Content uniformity (within the container)
• Chemical properties
– Assay
– Degradation products
– Content preservatives
– Degradation preservatives
– Content antioxidants
• Microbial properties
– Microbial purity
• Container closure system properties
– Functionality tests
General requirements for COPP Application:
1. A forwarding letter/application shall be addressed to DDC(I)/ADC(I) of respective CDSCO zonal/sub zonal offices with copy of covering letter & product summary sheet to DCG(I) (WHO-cell) by authorized person only.
2. The forwarding letter/application shall be accompanied with List of products applied for grant of COPP, along with the product permission copy (manufacturing licence issued by the SLA) & notarized product summary sheet, site master file as per WHO-GMP requirement.
|
S. No. |
Name |
Number of batches produced in last two years (with scale R&D/Pilot/ Commercial) |
Stability studies (maximum period completed) in months Accelerated / Real time |
Process Validation |
Analytical Method Validation |
Cleaning Validation |
Annual Product Review |
If permitted |
|||||||||||
|
Completed/ |
Completed |
Completed |
Completed |
||||||||||||||||
|
Acc |
R. T. |
||||||||||||||||||
|
1 |
Example Tablet |
20 (Commercial) |
6 M |
36 M |
Completed |
Completed |
Completed |
Not Completed |
Y |
||||||||||
3. Manufacturing layout (it is preferred if men and material flow, pressure flow drawing are also given)
4. HVAC schematics and details of areas (Where in clearly specify the filtration level & classification of core areas & rooms as required in section 3.3 of SMF) and Water system – Schematic diagrams along with the components.
5. List of personnel (with designation, qualification & experience), List of equipments, instruments, utilities along with make and model & capacity.
6. List of primary & secondary Impurity and Reference standards/cultures available with the firm (relevant to the applied products for grant of COPP).
Model Certificate of a Pharmaceutical Product
Certificate of a Pharmaceutical Product1
This certificate conforms to the format recommended by the World Health Organization (general instructions and explanatory notes attached).
No. of Certificate:
Exporting (certifying) country:
Importing (requesting) country:
1. Name and dosage form of product:
1.1 Active ingredient(s)2 and amount(s) per unit dose:3
For complete qualitative composition including excipients, see attached4.
1.2 Is this product licensed to be placed on the market for use in the exporting country?5 Yes/No (key in as appropriate)
1.2 Is this product actually on the market in the exporting country? Yes/no/unknown (key in as appropriate)
If the answer to 1.2 is yes, continue with section 2A and omit section 2B.
If the answer to 1.2 is no, omit section 2A and continue with section 2B.6
2A.1 Number of product licence7 and date of issue:
2A.2 Product-licence holder (name and address):
2A.3 Status of product-licence holder:8 a/b/c (key in appropriate category as defined in note 8)
2A3.1 For categories b and c the name and address of the manufacturer producing the dosage form are: 9
2A.4 Is Summary Basis of Approval appended?10
yes/no (key in as appropriate)
2A.5 Is the attached, officially approved product information complete and consonant with the form are: 9 Yes/no/not provided (key in as appropriate)
2A.6 Applicant for certificate, if different from licence holder (name and address):12
2B.1 Applicant for certificate (name and address):
2B.2 Status of applicant: a/b/c (key in appropriate category as defined in note 8)
2B2.1 For categories b and c the name and address of the manufacturer producing the dosage form are: 9
2B.3 Why is marketing authorization lacking?
Not required/not requested/under consideration/refused (key is as appropriate)
2B.4 Remark: 13
3. Does the certifying authority arrange for periodic inspection of the manufacturing plant in which the dosage form is produced?
Yes/no/not applicable14 (key in as appropriate If no or not applicable proceed to question 4.
3.1 Periodicity of routine inspections (years):--
3.2 Has the manufacture of this type of dosage form been inspected? Yes/no (key in as appropriate) Do the facilities and operations conform to GMP as recommended by the World Health Organization? 15 Yes/no (key in as appropriate)
4. Does the information submitted by the applicant satisfy the certifying authority on all aspects of the manufacture of the product? 16
Yes/no (key in as appropriate)
If no, explain:
Address of certifying authority:
Telephone number: ----------------------------------
Fax number: --------------------------------------
Name of authorized person:
Signature:
Stamp and date:
General Instructions Please refer to the guidelines for full instructions on how to complete this form and information on the implementation of the Scheme. The forms are suitable for generation by computer. They should always be submitted as hard copy, with responses printed in type rather than handwritten. Additional sheets should be appended, as necessary, to accommodate remarks and explanations.
Explanatory notes
1. This certificate, which is in the format recommended by WHO, establishes the status of the pharmaceutical product and of the applicant for the certificate in the exporting country. It is for a single product only since manufacturing arrangements and approved information for different dosage forms and different strengths can wary.
2. Use, whenever possible, International Nonproprietary Name (INNs) or national nonproprietary name.
3. The formula (complete composition) of the dosage form should be given on the certificate or be appended.
4. Details of quantitative composition are preferred, but their provision is subject to the agreement of the product-licence holder.
5. When applicable, append details of any restriction applied to the safe, distribution or administration of the product that is specified in the product licence.
6. Section 2A and 2B are mutually exclusive.
7. Indicate, when applicable, if the licence is provisional, or the product has not yet been approved.
8. Specify whether the person responsible for placing the product on the market:
(a) Manufactures the dosage form;
(b) Packages and/or labels a dosage form manufactured by an independent company; or
(c) Is involved is none of the above.
9. This information can be provided only with the consent of the product-licence holder or, in the case of non-registered products, the applicant. Non-completion of this section indicates that the party concerned has not agreed to inclusion of this information. It should be noted that information concerning the site of production is part of the product licence. If the production site is changed, the licence must be updated or it will cease to be licence.
10. This refers to the document, prepared by some national regulatory authorities, that summarizes the technical basis on which the product has been licensed.
11. This refers to product information approved by the competent national regulatory authority, such as a Summary of Product Characteristics (SPC).
12. In this circumstance, permission for issuing the certificate is required from the product-licence holder. This permission must be provided to the authority by the applicant.
13. Please indicate the reason that the applicant has provided for not requesting registration:
(a) the product has been developed exclusively for the treatment of conditions-particularly tropical diseases-not endemic in the country of export;
(b) the product has been reformulated with a view to improving its stability under tropical conditions;
(c) the product has been reformulated to exclude excipients not approved for use in pharmaceutical products in the country of import;
(d) the product has been reformulated to meet a different maximum dosage limit for an active ingredient;
(e) any other reason, please specify.
14. Not applicable means that the manufacture is taking place in a country other than that issuing the Product certificate and inspection is conducted under the aegis of the country of manufacture.
15. The requirements for good practices in the manufacture and quality control of drugs referred to the Certificate are those included in the thirty-second report of the Expert Committee on Specifications for Pharmaceutical Preparations (WHO Technical Report Series, No. 823, 1992, Annex 1). Recommendations specifically applicable to biological products have been formulated by the WHO Expert Committee on biological Standardization (WHO Technical Report Series, No. 822, 1992, Annex 1).
16. This Section is to be completed when the product-licence holder or applicant conforms to status (b) or (c) as described in note 7 above. It is of particular importance when foreign contractors are involved in the manufacture of the product. In these circumstances the applicant should supply the certifying authority with information to identify the contracting parties responsible for each stage of manufacture of the finished dosage form, and the extent and nature of any controls exercised over each of these parties.
The Layout for this Model Certificate is available on WordPerfect from the Division of Drug Management and Policies, World Health Organization, 1211 Geneva 27, Switzerland.
ACTD Format for Application in Asean Countries:
Permission to conduct BE Studies for Export:
Documents to be submitted for grant of permission to conduct Bioequivalence studies for export purpose.
A large number of applications are being filed to the office of DCG (I) at CDSCO (HQ) by Pharmaceutical companies, both manufacturers and importers as well as CRO’s on behalf of them, requesting for the approval to carry out BE studies with various pharmaceutical dosage formulations on Indian subjects.
In light of the above, for easy processing of such applications and to bring uniformity in decision making all stake holders of therefore mentioned activities are hereby advised to submit their applications with following documents. All applications should accompany the documents with proper index & page number.
Requirements for BE study of a new molecule not approved in India but approved in the other countries.
1. Application in Form-44 duly signed, by the competent authority with name and designation.
2. Treasury Challan of Rs. 25000/- as per Drugs & Cosmetic Rules.
3. Undertaking by the Principal Investigator (PI) as per appendix VII of schedule “Y” of Drugs and Cosmetic Rules.
4. A copy of the approval granted to the BE study centre by CDSCO.
5. Sponsor’s Authorization letter duly signed by the competent authority on their letterhead.
6. The study protocols.
7. The study synopsis
8. Pre-clinical single dose data and repeated dose toxicity data.
9. Clinical study data and published report of pharmacokinetic and pharmacodynamic study carried out in healthy volunteers/patients data published in reputed journals.
10. Regulatory status of the drug.
11. Names of the countries where the drug is currently being marketed (to be mentioned in the covering letter also).
12. Package literature on the international product
13. Complete Certificate of Analysis of same batches (both test & reference formulations) to be used in the BE study.
14. In the case of multiple dose BE study adequate supporting safety data should be submitted.
15. In the case of Injectable preparation the sub-acute toxicity should be submitted on the product of the sponsor, generated in two species for adequate duration.
16. Depending on the nature of the drug like cytoxic agent, hormonal preparations etc. Proper justification for conducting studies on healthy volunteers/patients or male/ female should be submitted.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
New Drugs approved in India within period of 1 year:-
1. Application in Form-44 duly signed, by the competent authority with name and designation.
2. Treasury Challan of Rs. 25000/- as per Drugs & Cosmetic Rules.
3. Undertaking by the Principal Investigator (PI) as per appendix VII of schedule “Y” of Drugs and Cosmetic Rules.
4. A copy of the approval of the BE study centre from CDSCO.
5. Sponsor’s Authorization letter duly signed by the competent authority on their letterhead.
6. The study protocols.
7. Clinical study data and published report of pharmacokinetic and pharmacodynamic study carried out in healthy volunteers data published in reputed journals.
8. Package literature on the international product.
9. Complete Certificate of Analysis of same batches (both test & reference formulations) to be used in the BE study.
10. In the case of multiple dose BE study adequate supporting safety data should be submitted.
11. In the case of Injectable preparation the sub-acute toxicity should be submitted on the product of the sponsor, generated in two species for adequate duration.
12. Depending on the nature of the drug like cytoxic agent, hormonal preparations etc. Proper justification for conducting studies on healthy volunteers/patients or male/ female should be submitted.
New Drugs approved within period of more than 1 year & less than 4 years:-
1. Application in Form-44 duly signed, by the competent authority with name and designation
2. Treasury Challan of Rs. 15000/- as per Drugs & Cosmetic Rules.
3. Undertaking by the Principal Investigator (PI) as per appendix VII of schedule “Y” of Drugs and Cosmetic Rules.
4. A copy of the approval of the BE study centre from CDSCO.
5. Sponsor’s Authorization letter duly signed on their letterhead by the competent authority.
6. The study protocols.
7. Complete Certificate of Analysis of same batches (both test & reference formulations) to be used in the BE study.
8. In the case of multiple dose BE study adequate supporting safety data should be submitted.
9. In the case of Injectable preparation the sub-acute toxicity should be submitted on the product of the sponsor, generated in two species for adequate duration.
10. Depending on the nature of the drug like cytoxic agent, hormonal preparations etc. Proper justification for conducting studies on healthy volunteers/patients or male/ female should be submitted.
BE NOC for all the drug products in modified release form irrespective of their approval status:-
1. Application in Form-44 duly signed, by the competent authority with name and designation
2. Treasury Challan of Rs. 15000/- as per Drugs & Cosmetic Rules.
3. Undertaking by the Principal Investigator (PI) as per appendix VII of schedule “Y” of Drugs and Cosmetic Rules.
4. A copy of the approval of the BE study centre from CDSCO.
5. Sponsor’s Authorization letter duly signed on their letterhead by the competent authority.
6. The study protocols.
7. Complete Certificate of Analysis of same batches (both test & reference formulations) to be used in the BE study.
8. In the case of multiple dose BE study adequate supporting safety data should be submitted.
9. In the case of Injectable preparation the sub-acute toxicity should be submitted on the product of the sponsor, generated in two species for adequate duration.
10. Depending on the nature of the drug like cytoxic agent, hormonal preparations etc. Proper justification for conducting studies on healthy volunteers/patients or male/ female should be submitted.
All above requirements are general in nature, however depending on the nature of the drug, disease and studies further specific information may also be required to be furnished by the firm.
Regulatory Control of Pharmacovigilance System
Pharmacovigilance is the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other possible drug-related problems. The scope of the Pharmacovigilance has been to:
* improve patient care and safety in relation to the use of medicines and all medical and paramedical interventions
* improve public health and safety in relation to the use of medicines
* contribute to the assessment of benefit, harm, effectiveness and risk of medicines, encouraging their safe, rational and more effective (including cost-effective) use
* promote understanding, education and clinical training in Pharmacovigilance and its effective communication to the public.
Recently, its concerns have been widened to include herbals, traditional and complementary medicines, blood products, biologicals, medical devices and vaccines. Pharmacovigilance also concerns substandard medicines, medication errors, lack of efficacy reports, use of medicines for indications that are not approved and for which there is inadequate scientific basis, case reports of acute and chronic poisoning, assessment of drug-related mortality, abuse and misuse of medicines and adverse interactions of medicines with chemicals, other medicines, and food.
Importance of Pharmacovigilance:
It highlights the need for critical examination of the Strengths and weakness of present Pharmacovigilance systems in order to increase their impact. It anticipates developments necessary to meet the challenges of the next ten years. It argues that the distinctive approaches adopted by different countries in response to their individual needs should be supported and fostered. The document also highlights the importance of collaboration and communication at local, regional and international levels, to ensure Pharmacovigilance delivers its full benefits.
CONSTITUTES OF THE PHARMACOVIGILANCE PROCESS
Pharmacovigilance process starts at the way beginning of clinical development process of a drug product and continues throughout the life cycle of the product. Broadly it can be divided into two phases:
1) Pre Marketing Pharmacovigilance Process
2) Post Marketing Pharmacovigilance Process
Pre Marketing Pharmacovigilance Process:
The assessment of clinical data and Safety and Efficacy data during the drug product development known as the pre marketing Pharmacovigilance process. It involves the measuring of the adverse drug reactions in whole the phases of the clinical trials. On the basis of these data or case report form informs the agencies and manage the risk benefits ratio of the drug product. During the clinical data the adverse drug reaction occur are known as the expected adverse drug reaction and those which are comes during the post marketing Pharmacovigilance process are known as the unexpected adverse drug reaction.
Post Marketing Pharmacovigilance Process:
Phase 2nd of the Pharmacovigilance process after a medicines gets launched in the market and until the time that it remain in the market is the post approval Pharmacovigilance process. It is the post marketing Pharmacovigilance process that’s the regulatory agencies of the world vigilant about the reporting of adverse events happens spontaneously.
Pharmacovigilance in different countries/regions
All the regions of the world have their own particular Pharmacovigilance system, through based on WHO guidelines.
Pharmacovigilance in Europe
Pharmacovigilance system in Europe is coordinated by the European Medicines Agency (EMA) and conducted by the National Competent Authorities (NCAs). The EMA maintains and develops the Pharmacovigilance database comprising all suspected serious adverse drug reaction observed in the European region. Here, the Pharmacovigilance system is called EUDRA Vigilance and contains separate but similar database of human and veterinary reactions. EMA Pharmacovigilance legislation regulated by Article 106 of Directive 2001/83/EC, Directive 2001/20/EC & Article 26 of Regulation (EC) No. 726/2004 EMEA& EC
Pharmacovigilance in United States
Here Pharmacovigilance has a multi faced approach. Three branches of the Pharmacovigilance in the USA has been defined by the FDA to evaluate product risks and promote the safe use of products by the American people. These three division / branches comes in the office of Surveillance and epidemiology (OSE).
Three Divisions within OSE:
1. Division of Drug Risk Evaluation (DDRE)
2. Division of Medication Errors and Technical Support (DMETS)
3. Division of Surveillance, Research and Communication Support (DSRCS)
In United State the Pharmacovigilance Legislation Regulated 21 CFR 314.80, 314.98 FDA, CDER, CBER.[7]
Pharmacovigilance in India
The central drugs Standard control organisation (CDSCO), ministry of health and family welfare, Govt of India launched the national Pharmacovigilance programmed (NPP) in November, 2004 based on the WHO recommendations made in the document titled” safety monitoring of Medicinal products-guidelines for setting up and Running a Pharmacovigilance Centre” the whole country is divided into zones and regions for the operational efficiency, CDSCO, new Delhi is at the top of the hierarchy by two zonal Pharmacovigilance centre viz. seth GS medical college , Mumbai and AIIMS, New Delhi.
Guidelines for Analytical Method Validation:
INTRODUCTIONS:
The objective of validation of an analytical procedure is to demonstrate that it is suitable for its intended purpose.
Analytical procedures used to measure the quality of pharmaceutical products span almost the entire range of currently available technologies and techniques.
It is important to keep in mind that the most important aspect of any analytical method is the quality of the data it ultimately produces.
The analytical procedure refers to the way of performing the analysis. It should describe in detail the steps necessary to perform each analytical test. This may include, but is not limited to, the sample, the reference standard and the reagents preparations, use of the apparatus, generation of the calibration curve, use of the formulae for the calculation, etc.
All relevant data collected during validation and formulae used for calculating validation characteristics should be submitted and discussed as appropriate. Well-characterized reference materials, with document purity, should be used throughout the validation study. The degree of purity depends on the intended use.
TYPES OF ANALYTICAL PROCEDURE TO BE VALIDATED:
The Discussion of the validation of analytical procedure is directed to the four most common types of analytical procedures:
Identification tests :
Tests are intended to ensure the identity of analyte in a sample this is normally achieved by comparison of a property of the sample (e.g. spectrum, chromatographic behavior chemically reactivity, etc) to that of a reference sample.
Quantitative tests for impurities content:
For the impurity in a sample either test is intended to accurately.
Limit tests for the control impurities:
Different validation characteristics are required for a quantitative test than for a limit tests.
Quantitative tests of the active tests moiety in sample of drug substance or drug product or other selected component(s) in the drug product.
The assay represents a quantitative measurement of major component (s) applies when assaying for the active or selected component(s). the same validation characteristic may also apply to assays associated with other analytical procedure (e.g., dissolution).
The objective of the analytical procedure should be clearly understood since this will govern the validation characteristic which needs to be evaluated. Typical validation characteristic which should be considered are listed below:
* Accuracy.
* Precision.
* Repeatability.
* Intermediate precision.
* Reproducibility.
* Specificity.
* Detection limit.
* Quantitation Limit.
* Linearity.
* Range.
* Robustness
The degree of revalidation required depends on the nature of the changes. Certain other changes may require validation as well.
|
Type of Analytical Procedure |
Identification |
Testing for Impurities |
Assay Dissolution(Measurement Only) Content /Potency |
|
|
Characteristics |
Quantitative |
Limit |
||
|
Accuracy |
- |
+ |
- |
+ |
|
Precision |
|
|
|
|
|
Repeatability |
- |
+ |
- |
+ |
|
Intermediate Precision |
- |
+(1) |
- |
+(1) |
|
Specificity(2) |
+ |
+ |
+ |
+ |
|
Detection Limit |
- |
-(3) |
+ |
- |
|
Quantitation Limit |
- |
+ |
- |
- |
|
Linearity |
- |
+ |
- |
+ |
|
Range |
- |
+ |
- |
+ |
|
(-): Signifies that this characteristic is not normally evaluated. (+): Signifies that this characteristic is normally evaluated. (1):in cases where reproducibility (see glossary) has been performed, intermediate precision is not needed. (2) lack of specificity of one analytical procedure could be compensated by other supporting analytical procedure(s) (3) may be needed in some cases |
||||
ANALYTICAL PERFORMANCE CHARACTERISRTICS:
SPECIFICITY:
Specificity is the ability to assess unequivocally the analyte in the presence of components which may be expected to be present. Typically, these might include impurities, degradants, matrix, etc. Lack of specificity of an individual analytical procedure may be compensated by other supporting analytical procedure(s). Non-Numerical data (Raw Data, Chromatograms).
Comparison of responses from samples with and without analyte present.
Identification:
Identification tests should be able to discriminate between compounds of closely related structure which are likely to be present.
Identification test may be applied to maintain structurally similar to or closely related to the analyte to confirm that a positive response is not obtained.
Assay& Impurity Test (s):
The method was intended to detect trace quantities of the active ingredients for purpose of a cleaning validation the detection and quantification limits are appropriate and necessary. Validation of each assay or test method should be performed on a case basis, to ensure that the parameter is appropriate for the methods intended to use.
The approach is similar for both assay and impurity tests:
Impurities are Available :
Demonstration of the discrimination of the analyte in the presence of impurities and or excipients; Practically, this can be done by spiking pure substance with appropriate levels of impurities and/or excipients and demonstrating that the assay result in unaffected by the presence of these materials. Impurity test, the discrimination may be established by spiking drug substance or drug product with appropriate level of impurities and demonstrating the separation of these impurities individually and /or from other components in the sample matrix./or from other components in the sample matrix.
Impurities are not Available :
If impurity or degradation product standard are unavailable ,specificity may be demonstrated by comparing the tests results of sample containing impurities or degradation product to a second well – characterized procedure.
E.g. Pharmacopoeial method or other validation analytical procedure. As appropriate, this should include samples stored under relevant stress condition: light, heat, humidity, acid/base hydrolysis and oxidation,
• For the assay, the two results should be compared.
• For the impurity tests, the impurity profile should be compared.
Peak purity tests may be useful to show that the analyte chromatographic peak is not attributable to more than one component (e.g., diode array, mass spectrometry).
To the drug formulation were added known amount of API was estimated by the prescribed method to know it.

The values suggest that the method is specific for estimation of API and amounts of other excipients do not have any impact on estimation of the analyte.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Recommended Data:
LINEARITY:
The linearity of an analytical procedure is its ability (within a given range) to obtain test results which are directly proportional to the concentration (amount) of analyte in the sample.
It may be evaluated by visual inspection of a plot of signals as a function of analyte concentration or content. If there is linear relationship, tests results should be evaluated by appropriate statistical method.
Linearity should be evaluated by visual inspection of plot of signals as function of analyte concentration or content. If there is linear relationship, test result should be evaluated by appropriate statistical methods.
Linearity should be determined concurrently during the accuracy study .Classical linearity acceptance criteria are 1) that the correlation coefficient of the linear regression lines is not more than some number close to 1, and 2) that the y- intercept should not differ significantly from zero.
When linear regression analyses are performed, it is important not to force the origin as (0, 0) in the calculation. This practice may significantly skew the actual best-fit slope through the physical range of use.
Visual inspection of standard plot.
Correlation coefficient.
Y-intercept.
Slope.
Residual sum of squares.
Name of Ingredient: (I1): API
RANGE:
The range of an analytical procedure is the interval between the upper and lower concentration (amounts) of analyte in the sample (including these concentrations) for which it has been demonstrated that the analytical procedure has a suitable level of precision, accuracy and linearity.
Range is normally derived from linearity studies and depends on the intended application of the procedure. It is established by confirming that the analytical procedure provides an acceptable degree of linearity, accuracy and precision when applied to samples containing amounts of analyte within or at the extremes of the specified range of the analytical procedure. The following minimum specified ranges should be considered.
ASSAY : 70% to 130%
CONTENT UNIFORMITY: Minimum of 70 %to 130%
DISSOLUTION : Not less than 70% of label claim.
IMPURITY : Reporting level of an impurity to 120 %
NOTE: for the validity of impurity test procedure carried out during development.
If assay and purity are performed together as one test and only a 100% standard is used. Linearity should cover the range from the reporting level of the impurities1 to 120 % of the assay specification.
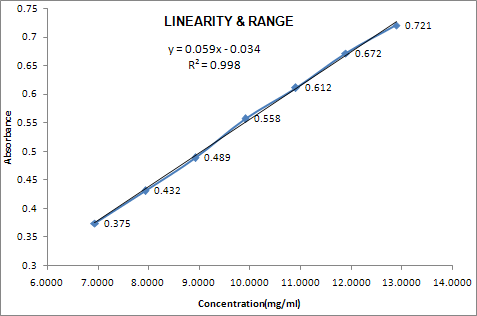
GRAPHICAL REPRESENTATION:
|
S. No. |
Conc. Level (%) |
Conc. (mcg/ml) |
Response (AREA/Abs.) |
|
1 |
70 |
6.9427 |
0.375 |
|
2 |
80 |
7.9345 |
0.432 |
|
3 |
90 |
8.9263 |
0.489 |
|
4 |
100 |
9.9181 |
0.558 |
|
5 |
110 |
10.9099 |
0.612 |
|
6 |
120 |
11.9017 |
0.672 |
|
7 |
130 |
12.8936 |
0.721 |
|
Correlation coefficient |
0.99939 |
||
|
Returns the square |
0.99878 |
||
|
INTERCEPT |
-0.03 |
||
|
SLOPE |
0.06 |
||
Recommended Data:
ACCURACY
The closeness of agreement between the value which is accepted either as a conventional true value or an accepted reference value, and the value found.
Note: When measuring accuracy, it is important to spike placebo preparations with varying amounts of active ingredient(s). If a placebo cannot be obtained, then a sample should be spiked at varying levels. In both cases, acceptable recovery must be demonstrated.
Direct comparison of your test results with the known results.
Direct comparison of your test results with the results from a second method.
Comparison of your spiked recovery results to 100% recovery.
Infer accuracy from precision, linearity, and specificity.
ASSAY:
Drug Substance:
A. Application of an analytical procedure to an analyte of known purity (e.g. reference material);
B. Comparison of the results of proposed analytical procedure with those of a second well Characterized procedure the accuracy of which is stated and/or defined (independent Procedure see 3.12).
C. Accuracy may be inferred once precision, linearity and specificity have been established.
Drug Product:
Application of the analytical procedure to synthetic mixture of the drug product components to which known quantities of the drug substance to be analysed have been added.
In case where it is impossible to obtain sample of the drug product components. It may be acceptable either to add known quantities of the analyte to the drug product or to compare the result obtained from a second, well characterized procedure, the accuracy of which is stated and/or defined (independent Procedure, see3.12.).
Accuracy may be inferred once .precision, linearity and specificity have been may inferred once precision, linearity and specificity have been Established.
IMPURITIES (Quantitation)
Accuracy should be assessed on the sample (drug substance /drug product) spiked with known amount of impurities. In case where it is impossible to obtain sample of certain impurities .and /or degradation product. The response factor of the drug substance can be used.
It should be clear how the individual or total impurities are to be determined. e.g.: weight /weight or area percent, in all cases with respect to the major analyte.
RECOMMENDED DATA:
Accuracy should be assessed using a 9 determination over a minimum of 3 concentration level covering the specified range (e.g. 3 concentration /3 replicates each of the total analytical procedure).
Accuracy should be reported as percent recovery by the assay of known added of Analyte in the sample .or difference between the mean the accepted true valve .together with confidence Intervals.
Summary Data:
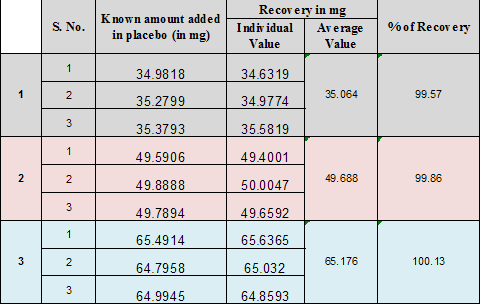
Recommended Data:
Precision:
The precision of an analytical procedure expresses the closeness of agreement (degree of scatter) between a series of measurements obtained from multiple sampling of the homogeneous sample under the prescribed conditions. Precision may be considered at three levels: repeatability, intermediate precision and reproducibility.
The precision of an analytical procedure is usually expressed as the variance, standard deviation or coefficient of variation of a series of measurements.
* Standard Deviation
* Relative Standard Deviation (coefficient of variation)
* Confidence Interval
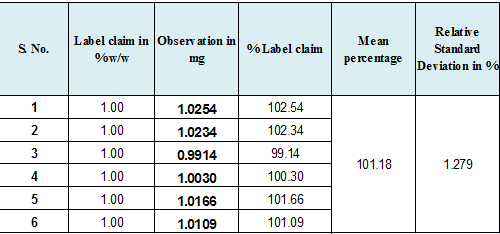
Recommended Data:
Repeatability:
Repeatability expresses the precision under the same operating condition over a short interval of time.
Repeatability is also termed intra –assay precision a minimum of 9 determinations covering the specified range for the procedure (e.g.-3 concentration/3 replicates each) or a minimum of 6 determinations at 100% of the tests concentration.
Intermediate Precision:
Intermediate precision express within –laboratories variations: different days, different analysis, different analysis, different equipment etc.
The applicant should establish the effect of random events on the precision (six replicate) of the analytical procedure. Typical variation to be studied include days, analyst, equipment, etc It is not considered necessary to studied these effect individually. The use of an experimental design (matrix) is encouraged.
The intermediate precision of an analytical procedure is usually expressed as the variance, standard deviation or coefficient of variation of a series of measurements of both analyte.
* Standard Deviation
* Relative Standard Deviation (coefficient of variation)
* Confidence Interval
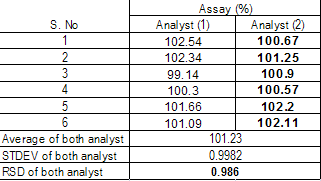
Recommended Data:
Reproducibility:
Reproducibility expresses the precision between laboratories (collaborative studies usually applied to standardization of methodology).
Reproducibility should be considered in case of the standardization of an analytical procedure ,for the Instance, for indusion of procedures in pharmacopoeias.
Detection Limit:
The detection limit of an individual analytical procedure is the lowest amount of analyte in a sample, Which can be detected but not necessary quantified as an extract valve. Several approaches for determining the detection limit are possible, depending on whether the Procedure is a non-instrumental or instrumental, Approaches other than those listed below may be accepted.
Based On Visual Evaluation:
Visual evaluation may be used for non-instrumental method but may also be used with instrumental methods. The detection limit is determined by the analysis of sample .with known concentration of analyte and By establishing the minimum level at which the analyte can be reliably detected.
Based On Signal To Noise:
This approach can only be applied to analytical procedure which exhibit base line noise. Determination of the signal- to noise ratio is performed by comparing measured signal from Sample with known low concentration of analyte with those of blank sample and establishing the minimum concentration at which the analyte can be reliably detected. A signal-to noise ratio Between 3 or 2:1 is generally considered acceptable for detection limit.
Based On The Standard Deviation Of The Response And The Slope:
The detection limit (DL) may be expressed as:
DL = 3.3 σ/S
Where σ = the standard deviation of the response
S = the slope of calibration curve
The slope S may be estimated from the calibration curve of the analyte .The estimate of S may be carried out in variety of ways ,for example :
Based On The Standard Deviation Of The Blank:
Measurement of the magnitude of the analytical background response is performed be analyzing an appropriate number of blank sample and calculating the standard deviation of these response.
BASED ON THE CALIBRATION CURVE:
A specific calibration curve should be studied using sample containing an analyte in the range of Sample containing an analyte in the range of DL. The residue standard deviation of a regression Line or the standard deviation of y- intercepts of regression lines may be used as the standard Deviation.
RECOMMENDED DATA:
The detection limit and the method used for determining the detection limit should be presented. If DL is determined based on visual evaluation or based on signal on noise ratio, the presentation Of relevant chromatograms is considered acceptable for justification.
Quantitation Limit:
The Quantitation limit of an individual analytical procedure is the lowest amount of analyte in a sample which can be quantitatively determined with suitable precision and accuracy. The Quantitation limit is a parameter of quantitative assays for low levels of compounds in sample matrices, and is used particularly for the determination of impurities and/or degradation products.
BASED ON THE VISUAL EVALUATION:
Visual evaluation may be used for non- instrumental method but may also be used with instrument method. The Quantitation limit is generally determined by the analysis of sample of known concentration of analyte and by establishing the minimum level at which the analyte can be quantified With acceptable accuracy and precision.
BASED ON SIGNAL TO NOISE :
This approach can only be applied to analytical procedure that exhibit base line noise Determination of the signal-to-noise ratio is performed by comparing measured signal from sample with known low concentration of analyte with those of blank samples and by establishing the minimum concentration at which the analyte can be reliably quantified. A typical signal- to- noise ratio is 10:1
BASED ON THE STANDARD DEVIATION OF THE RESPONSE AND THE SLOPE :
The Quantification limit (QL) may be expressed as:
QL =10 σ/S
Where σ = the standard deviation of the response
S = the slope of the calibration curve slope
The slope S may be estimated from the calibration curve of the analyte. The estimate of S may be Carried out in a variety of ways ,for example :
Based On The Standard Deviation Of The Blank:
Measurement of the magnitude of analytical background response is performed by analyzing an Appropriate number of blank samples and calculating the standard deviation of these responses.
Based On The Calibration Curve:
A specific calibration curve should be studied using sample containing an analyte in the range of QL. The residue standard deviation of a regression line or the standard deviation of y-intercept Regression line may be used as the standard deviation.
Recommended Data:
The detection limit and the method used for determining the Quantitation limit should be presented If QL is determining based on visual evaluation or based on signal to noise ratio ,the presentation of the relevant chromatogram is considered acceptable for justification.
ROBUSTNESS:
The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate, variations in method parameters and provides an indication of its reliability during normal usage.
Note: Ideally, robustness should be explored during the development of the assay method.
The actual method validation will ensure that the final, chose range is robust.
The evaluation of robustness should be considered during the development phase and depends on the type of procedure under study. once consequence of evaluation of robustness should be that a series of system suitability parameter.(e.g. ;resolution test) is established to ensure that the validity of analytical procedure is maintained whenever is used. Example of typical variation is Stability of analytical solution.
Extraction time.
In the case of liquid chromatography, example of typical variation is:
01. Influence of variation of pH in a mobile phase
02. Influence of variation in mobile phase composition, different columns (different lots and/or suppliers).
03. Temperature.
04. Flow rate.
In the case of gas chromatography, example of typical variation is:
01. different columns (different lots and/or supplier ),
02. Temperature.
03. Flow rate.
SYSTEM SUITABILITY:
Prior to the start of laboratory studies to demonstrate method validity, some type of system suitability must be done to demonstrate that the analytical system is performing properly.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Examples include:
Replicate injections of a standard preparation for HPLC and GC methods.
Standardization of a volumetric solution followed by assays using the same burette for titrimetric methods.
Replicate scanning of the same standard preparation during UV-VIS assays, etc.
When the method in question utilizes an automated system such as a chromatograph or an atomic absorption spectrophotometer, a suitable standard preparation should be intermittently measured during the sample analysis run. The responses generated by the standard should exhibit a reasonable relative standard deviation. This is done primarily to demonstrate the stability of the system during sample measurements. System suitability for dissolution studies should be performed using both USP Non- disintegrating and disintegrating tablets prior to the validation of dissolution method.
|
OBSERVATION SUMMARY SHEET |
||||
|
S. No. |
CONTENT |
OBSERVATIONS |
LIMITS |
|
|
1.0 |
Specificity |
For Permethrin sample was added in placebo the % recovery observed. Thus this has not any impact on the proposed method. |
99.57% to 100.13% |
98% to 102% |
|
2.0 |
Linearity & Range |
Linearity was studied by preparing standard solutions at different volume & the linearity range was found 6.9427mcg to 12.8936mcg |
Correlation Coefficient is 0.99939 |
Correlation Coefficient is NLT 0.997 |
|
3.0 |
Accuracy |
It was done by recovery studying using standard addition method known amount of Permethrin were added into the placebo and subjected them to the proposed HPLC method. |
The % recovery observed is 99.57% to 100.13% |
98% to 102 % |
|
4.0 |
Precision |
Six replicate samples were tested and the % RSD observed. |
1.279%% |
Less Than 2.0% |
|
5.0 |
Intermediate Precision |
Intermediate precision at inter day analysis as well as different HPLC system by another analyst and calculate the relative standard deviation of both analyst. |
The maximum deviation against first analyst is 0.986% |
Less Than 2.0% |
|
Conclusion: |
On the basis of the above observation it is concluded that the analytical method (UV) taken for validation is validated for different strength of same formulation. |
|||
Multicountry Regulatory Filling Strategy:
Taking advantage of the Common Technical Document (CTD) defined by the International Conference on Harmonisation (ICH), biopharmaceutical companies can significantly improve the speed and efficiency of preparing multi-country regulatory submissions while reducing the risk of costly delays. A strategic, multinational regulatory approach greatly increases the chances of marketplace success for both new and existing pharmaceutical products during these difficult times—assuming sponsor have the right resources and local expertise to effectively develop and implement the strategy. Leveraging the CTD The CTD is the key to leveraging the benefits of multi-country filings. Based upon the ICH M4 guidelines set forth in “Common Technical Document for the Registration of Pharmaceutical for Human Use—Quality, Safety and Efficacy,”1 the CTD provides a standardized structure for regulatory submissions that is acceptable to all ICH members—the European Union, Japan and the US. Other countries, including Australia, New Zealand, Canada, India, China, Turkey, Serbia, Croatia, Bosnia-Herzegovina, Ukraine and Kazakhstan also have elected to accept the CTD for regulatory submissions. A similar submissions format—the ASEAN CTD (ACTD)—is being developed by ASEAN member countries including Brunei, Cambodia, Indonesia, Laos, Malaysia, Myanmar, Philippines, Singapore, Thailand and Vietnam.2 This format is closely aligned with the CTD guidelines and will further expand the utility of CTD-based dossiers. The implementation of the ACTD should also facilitate approval and marketing of pharmaceutical products in these countries.
The ongoing switch to electronic submissions also facilitates the multi-country submissions process. By July 2009, the European Medicines Agency (EMEA) will strongly recommend eCTD submissions;3 other electronic formats and paper will be the exception. For submissions via the Decentralized or Mutual Recognition Procedures, applicants are also encouraged to use the eCTD. The move to electronic submissions minimizes the logistics of filing paper submissions that add significant time and expense to multi-country filings.
Understanding Regional Differences in CTD Submissions
Although the CTD makes multinational filings easier, it is important to remember that regulatory submissions in the EU, the US and elsewhere in the world continue to have significant differences. For example, an EU CTD typically contains more summary data in the CTD modules, as opposed to the cross references to full reports typically seen in submissions to the US Food and Drug Administration. The fact that a product has been approved in one country or region does not guarantee its approval in others. A successful multi-country filing strategy requires close attention to local requirements, familiarity with previous regulatory decisions and knowledge of the country’s medical practices. At the same time, regulatory approval of a pharmaceutical product in a major market will often improve its chances for acceptance in other countries. In some countries, for example Australia and New Zealand (see below), a CTD-based product dossier that has been approved for marketing in the EU or the US may be eligible for accelerated review and faster market approval.
A company’s ability to understand and accommodate local regulatory differences will have a substantial impact on the success of its multi-country submissions strategy. Many pharmaceutical companies contemplating multinational filings find that they benefit from outside regulatory resources with specific knowledge of local and regional regulations to help them refine their multi-country strategy and position their regulatory submissions for approval in the targeted markets.
Developing an Effective Multi-Country Submissions Strategy
While a multi-country filing strategy is important to maximize product potential, it does not mean that submissions should be filed simultaneously in each country. The appropriate submission strategy depends upon a variety of factors, such as:
product indication, location of potential • patients and clinical practice
pricing and reimbursement issues•
sponsor size and resources •
whether the product is a novel therapy • or an established treatment expanding into new market analysis of market penetration and mar• ket share potential, as well as the current market leader manufacturing capacity and distribution • logistics.
Companies expanding into new countries or regions have additional issues to consider, such as unfamiliarity with regulatory differences and the common requirement that a sponsor have a local legal entity and designated responsible person in order to sell pharmaceutical products in a particular country. When targeting the EU, companies must decide whether to follow the Centralized Procedure (which is mandatory for specified products and indications) or the Decentralized/Mutual Recognition route for their submissions.4 For eligible products, the Centralized Procedure via EMEA provides the advantage of receiving a single authorization covering all 27 EU Member States, plus Iceland, Norway and Liechtenstein. The Decentralized and Mutual Recognition Procedures afford the opportunity to select specific Member States based upon product acceptance, ease of market access or market value.
The order and timing of multi-country filings can have a significant effect on whether a submission is reviewed and approved quickly or is subject to delays. Although every situation is unique, the following are examples of sound strategic approaches for three common multi-country filing scenarios:
Mid-sized and Large Multinational Biopharmaceutical Companies
With New Products
It is often advantageous for major biopharmaceutical companies to submit their initial marketing authorization applications in the US and the EU in parallel to maximize their return on investment during the patent protection period. These applications would typically be followed by a “second wave” of applications in other important markets such as Canada, Australia, New Zealand, China and India, which have implemented most or all of the applicable ICH/CTD guidelines.
The key to this strategy is to develop a robust core dossier that can be adapted to accommodate the differing requirements of FDA and EMEA. This approach greatly reduces duplication of effort. Consideration of exact requirements and expectations in second wave countries is important early in the process, to ensure an awareness of local regulations so appropriate adjustments can be made to the file, as required. This advanced strategic planning can significantly reduce the risk of regulatory delays by anticipating questions typically raised by the individual regulatory agencies, such as requests for ethnic data or the selection of a comparator.
Small Companies With Novel Therapies
Smaller biopharmaceutical companies usually seek initial regulatory approval for their products in their home countries, and then use the approved dossier as the basis for applications to other countries. Innovative therapies are of interest in every country, so small companies are encouraged to submit their products for approval elsewhere. Smaller companies also face greater financial risks if their applications fail, so they must be aware of region-specific requirements, standard therapeutic-area practices and existing reference products when preparing applications for new areas. Medical and regulatory experts with local knowledge of the targeted country or region can be vital in formulating submissions that have the greatest chances for success.
Mid-sized and Larger Companies Already in the EU Seeking to Expand Market Reach
Pharmaceutical companies with a portfolio of approved products in the EU can leverage their existing approvals to expand into non-EU European countries such as Croatia, Serbia, Ukraine and Russia. Recent dossiers in CTD format can be used for submission to these countries, although some countries require local language translations and country-specific documents. Products that have been on the market for a number of years may require an updated dossier—not only to facilitate assessments in new markets, but also to maintain support for the license in the existing markets. Experience has shown that a local presence and established relationships with regulatory authorities can be important for market success in these countries.
The Importance of Local Knowledge
In all of these scenarios, knowledge of regulatory differences and other country-specific requirements is crucial to the success of any multi-country submissions strategy. Familiarity with local differences in advance of preparing a submission can mean the difference between a smooth review process and significant delays or failures. Following are a few examples that illustrate the importance of understanding and anticipating unique local situations.
Australia
A non-Australian company wishing to register a new medicinal product in Australia must have a sponsor, located in Australia, responsible for the drug’s manufacture, supply and marketing. The sponsor must be registered with the Therapeutic Goods Administration (TGA) before any submissions can be made. Pre-submission meetings with TGA are strongly encouraged. In addition to standard (Category 1) submissions, Australia offers an abridged (Category 2) submission process for products that have already been approved and marketed in other “acceptable” countries. The review timeframe is 20 weeks shorter than the standard (Category 1) process—about one-third faster. However, the product must be identical to the one registered in the acceptable countries in formulation, directions for use and indications.
New Zealand
A shortened review process also is offered by New Zealand for products that have been approved and marketed in the US, Canada, Australia, the EMEA (Centralized Procedure) or the United Kingdom (National procedure5). The product must be identical in all aspects to the approved product, including manufacturing site, specifications, indications and warnings. The Extended Abbreviated Process is completed within 135 days, instead of an average of 450 days, and also offers the sponsor significant savings on fees.
Indonesia
In November 2008, Indonesia’s Ministry of Health issued a new regulation that requires foreign companies marketing drugs in Indonesia to manufacture them there. The regulation states that foreign-owned companies will not be permitted to register imported drugs for sale unless they have an Indonesia-based drug production facility. The regulation, which has not yet been implemented, gives pharmaceutical manufacturers two years to comply.
Malaysia
Only local companies can submit an application for drug registration. Foreign companies with no local presence in Malaysia must designate a locally established legal entity to be the Market Authorization Holder (MAH). The MAH is responsible for submitting the product application and ensuring the product’s quality, safety and efficacy. In addition, the country’s National Pharmaceutical Control Bureau (BPFK) only accepts web-based submissions.
Succeeding With Multinational Filings
Today’s highly competitive marketplace requires biopharmaceutical companies to maximize the value of their products by shortening time to approval and launching the products in strategic markets as quickly as possible. A proactive, multi-country regulatory filing strategy helps biopharmaceutical companies of any size to accomplish both of those goals by increasing global sales, reducing regulatory submission costs and lowering the risk of regulatory delays or failures. To succeed with multinational registrations, a sponsor must:
identify key target markets for • submissions
understand important regional • differences
find the right local resources to avoid • regulatory pitfalls
create a robust CTD• develop a submissions strategy that • leverages the CTD to secure regulatory approvals in the shortest possible time.
A strategic multi-country approach will maximize the return on product investments while bringing important therapies to patients who need them around the world—a desirable outcome in any economic environment.
POSSIBLE BENEFITS:-
* Drug management functions are undergoing transformation in many countries as different ways of health sector reform are being implemented.
* Introduce the concept of governmental “regulation” and provide a framework for discussions about regulation in developing countries.
* Regulations and administrative guidelines are “enabled” by legislation enacted into constitutions or statutory law.
* Regulation of pharmaceuticals as a commodity (i.e., an article of commerce) has inherent contradictions
* Decentralization of drug regulatory authorities
* Licensing/GMP/QC
* Regulatory Changes regarding Drug Schedules
* Regulatory Changes regarding Facility Inspections
* Regulatory Changes regarding Advertising/Marketing
* Regulatory Changes regarding Accreditation of Facilities and Personnel
* Regulatory Changes regarding Pharmacovigilance
CONLUSION:-
Incorporating regulatory affairs into the pharmacycurriculum will make the pharmaceuticalprofessionals working in the area of regulatory affairs.
By their very nature, the existence of systems to regulate pharmaceuticals create costs and barriers to pharmaceutical availability. To protect the public health, however, access and quality of pharmaceuticals must be regulated. The means to accomplish this must be based on evidence of the effect of such regulation on availability and quality. It cannot be based on faith or belief.
Our present evidence base in this regard is very weak. To accomplish the laudable goal of basing pharmaceutical regulations on evidence of their effect on drug availability and quality, we need to be creative in studying existing regulatory systems and, in particular, we must pay attention to pharmaceutical availability and quality prior to, during, and after transition periods when the regulations undergo change(s). The World Health Organization and should coordinate and promote such studies.
REFERENCES:-
- International Conference on the Harmonisation of Technical Requirements for the Registration of Human Drugs. ICH Harmonised Tripartite Guideline Organisation of the Common Technical Document for the Registration of Pharmaceuticals for Human Use M4 (R3). 13 January 2004. Accessible at ich.org/cache/compo/276-254-1.html.
- “The ASEAN Common Technical Dossier (ACTD) for the Registration of Pharmaceuticals for Human Use. Organisation of the Dossier.” Accessible at hsa.gov.sg/publish/etc/medialib/hsa_library/health_prod-ucts_regulation/western_medicines/files_guidelines.Par.22449.File.dat/ACTD_OrganizationofDossier.pdf.
- “EMEA Implementation of Electronic-only Submissions and eCTD Submissions in the Centralised Procedure: Statement of Intent,” 22 January 2008, EMEA/563366/2007. Accessible at: www.emea.europa.eu/pdfs/human/regaffair/56336607en.pdf.
- Volume 2—Pharmaceutical Legislation Notice to applicants and regulatory guidelines medicinal products for human use. EudraLex. Accessible at ec.europa.eu/ enterprise/pharmaceuticals/eudralex/vol2_en.htm.
- New Zealand Regulatory Guidelines for Medicines, March 2009. Accessible at medsafe.govt.nz/regulatory/Guideline/Full%20-%20NZ%20Regulatory% 20Guidelines%20for%20Medicines.pdf.
- Gupta, P. K. 1998, "Development of Bhutan Medicines Act, 1998 and Bhutan Medicines Regulations, 1998", WHO: Regional Office for South East Asia, vol. WHO Project: BHU DAP 001,
- Pharmaceutical Inspection Cooperation Scheme (PICS) 1995, "Pharmaceutical Inspection Cooperation Scheme (PICS)", Pharmaceutical Inspection Cooperation Scheme (PICS) no. PIC/S/1/95, Available at: picscheme.org/docs/pdf/picschem.pdf.
- World Health Organization 1991, "The Public/Private Mix in National Health Systems and the Role of Ministries of Health", World Health Organization.
- World Health Organization 1993, "WHO Expert Committee on Specifications for Pharmaceutical Preparations", World Health Organ Tech Rep.Ser.33rd Report, vol. 834, pp. 1-30.
- World Health Organization 1995, Use of the WHO Certification Scheme on the Quality of Pharmaceutical Products Moving in International Commerce, WHO/DAP/94.21
- World Health Organization 1996, "Good pharmacy practice (GPP) in community and hospital pharmacy practice”, World Health Organization, vol. (unpublished WHO document WHO/PHARM/DAP 96.1).,
- World Health Organization 1998, How to Implement Computer-Assisted Drug Registration, World Health Organization, Geneva, Switzerland, WHO/DMP/RGS98.2.
- World Health Organization 1999a, "Effective Drug Regulation: what can countries do?", World Health Organization Technical Report Series, vol. WHO/HTP/EDM/MAC(11)/99.6,
- World Health Organization 1999b, Marketing Authorization of Pharmaceutical Products with Special Reference to Multisource (Generic) Products, WHO/DMP/RGS98.5 edn.
- World Health Organization 2000, Quality assurance of pharmaceuticals: A compendium of guidelines and related materials-Good Manufacturing practices and inspection, Volume 2.
- World Health Organization 2001, " Improving the quality and usefulness of drug regulatory authority websites." WHO Drug Information, vol. 15, no. 3/4, p. 82.
- World Health Organization 2001a, Good laboratory practice: training manual (TRAINER) , TDR/PRD/GLP/01.
- World Health Organization 2001b, Handbook: Good laboratory practice
- Quality practices for regulated non-clinical research and development, TDR/PRD/GLP/01
- World Health Organization. 2002a. The Importance of Pharmacovigilance. World Health Organization/Uppsala Monitoring Center ,pp. 1-48.
- World Health Organization 2002b, WHO's Ethical and Scientific Criteria for Pharmaceutical Advertising" , World Health Organization, Annex 3, DGO/ETHCDP/87.3, WHA21 .41.
- Pharmacovigilance;en.wikipedia.org/wiki/Pharmacovigilance.
- pharmaceutical-drugmanufacturers.com/articles/pharmacovigilance.html
- Careers in regulatory affairs from practitioner to professional, nature jobs biotechnology, Vol 20(4), page no. 409-410,april 2001
- ijprd.com
- sgs.com
- cdsco.nic.in
- emea.org
- fda.org
- ec.europa.eu/health/files/eudralex/vol-9/pdf/vol9_10-2004_en.pdf
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








