

{ DOWNLOAD AS PDF }
About Authors:
Emanual Michael Patelia*, Emma Spiking
University of Bedfordshire, England,
United Kingdom, LU1 3JU
ricky.emanual@gmail.com
Abstract
The new approach regarding the knocking down the protein expression by the use of shmRNA complimentary to the mRNA sequence of the respected protein expression have a major impact on the research area. The knockdown of P2X1 and ADRA1A genes have no effect on chromosomal DNA but have a lack of effect on expression of P2X1 and α1A receptors. The produced genetically engineered male mice can be of 100% infertile. The simple PCR (polymerase chain reaction) based approach can be used for the generation of knockdown cell lines. The Animals should have the treatments according to the guidelines mentioned under international society for applied ethnology. The SIRION biotech have shown the managed shRNA knockdown of about 90% significance interval.
REFERENCE ID: PHARMATUTOR-ART-2137
Introduction and rationale
The DNA sequence for the protein synthesis can be blocked or inhibit by the number of ways. The oligonucleotides specific towards the mRNA sequence of the respected protein is required for the inhibition of genetic expression. Thus binding of oligonucleotide towards the mRNA of the respected protein production does not produce the change in the chromosomal DNA but leads to a temporary alteration in gene expression referred to as transient knockdown. These can be done by three ways 1) blocking of transcription 2) degradation of mRNA transcript and 3) By blocking of either mRNA translation, pre-mRNA slicing sites, or nuclease cleavage. (summerton et al., 2007) (summerton et al., 1999).
The AhringerRNAi library by the use of organisms like C.elegans and drosophila provide the way of investigating gene function (Kamatin et al., 2007). The different means like “clustered regularly interspaced short palindromic repeats” can also be used in prokaryotic cells to silence the gene expression (Gilbert L.A. et al., 2013).
Another technology such as use of transcription activator liker effector nucleases (TALENS), that cab be used as plasmid or mRNA, that cleaves the non-homologus end joining to produce the DNA repair mechanism for the correction of cleavage. As the reparation includes the number of insertions and deletions at the required DNA sequence site (Sun N., et al., 2013).
Aims &Hypothesis
AIM: Use of Knockdown technique on expression of α1A adrenergic receptor and P2X1 receptor
Hypothesis:
- By deleting or inhibiting P2X1 and α1a receptors expression or genes, the male (present study on mice) become infertile
- Knockdown male can have the same sex drive or normal sexual activity.
- knockdown technique is more specific than other techniques(knock out technique)
Materials & Methods
Design and validation of shRNA sequences for knockdown
shRNA sequence for expression of P2X1 receptors
Mature miRNA sequence:
shRNA1: UGAGGUAGUAGGUUUGUAUAUU
shRNA2: UGAGGUAGAGGUUGUAUAGUU
shRNA3: UGAGGUAGUAGGUUGUAUAGUU
shRNA sequence for expression of ADRA1 receptors:
shRNA1: GCCAGGATTACAGTGTCCAAA
shRNA2: CCTGATTTCAAGCCCTCTGAA
shRNA3: CCTTTCAGAATGTCTTGAGAA
The expression of the receptors (P2X1) and (α1A) are major receptor proteins in vas deference muscle cell lines. The cell lines from the mouse vas deference cell lines can be used for the design of knockdown technique.The designing of 10, 15 or more shRNA sequence can be done by using in house algorithms. All that we are are using is the accession cDNA clone of the gene of interest. The cDNA clones should be identified for the P2X1 and ADRA1A genes.
The identification of shRNA sequences that produce potent gene gene silencing at single copy is one of the must carried steps in developing shRNA strain. One of the common method using PCR based strategy has been used at laboratory levels.
shRNA testing:
The choice of viral vector and cell typing for testing every shRNA is mainly dependent on target gene. This can be confirmed by antibiotic resistance.(puromycin, neomycin). There are two requirements that accurately validate the knockdown technique
1)Target cells that results into the expression of target protein and must be transduced by retro-or lenti virus under conditions that lead to a single genomic interaction of provirus and 2) An assay that allows the direct measurement of target proteins.
For testing the routinely used cells NIH3T3 Cells, they should be easily transduced with retrovirus and then cult ration of cells on the culture medium has to be done. Any mouse cells can be used for this kind of procedure. For the culturation of cells, the cell culture plates (10 cm BD Fallon) can be used.
Reagent setup: (Lukas E D., et al., 2012)
Reagents: ECO RI (20,000 ml-1, NEB, cat no. R0101), Xho1 (20,000U ml-1, NEB, cat no. R0146), Agel (5000 U ml-1, NEB, cat. No. R0552), Ncol (10,000 Uml-1, NEB, cat. No., R0193, T4 DNA ligase (20,000,000 Uml-1, NEB cat. No. M0290), calf intestinal phosphate (CIP 10,000 U ml-1, NEB, cat. No., M0290, platinum pfx DNA polymerase (2.5 Uml, invitrogen, cat. No., 1708-0219)
PCR tubes per plates 0.2 ml, PCR hood (DNA clean), DNA clean area, PCR thermal cycler with ramp increment mode, cell culture incubator (37 °C or regulator) cell culture laminar floe hood, bacterial incubator.
Oligonuceotides for PCR cloning: 97-mer oligos, MW 30,000 Da, 4nmol- 120ng, 4 nmol of oligonucleotides has to be resuspend in 120 µl of H2O to make a 1mg/ml stock. Then serial dilutions from the stocks can be made in ddH2O to a resultant concentration of 0.02 ng/µl. storage must be done at -20 °C.
Annealing Buffer: 5x for 1 ml of annealing buffer, combination of 500 µl 1 M pottasium acetate and 300 µl 0.5 M HEPES-KOH (pH 7.5), 20 µl 0.5 M magnesium acetate and 180 µl of ddH2O. The storage should done at room temperature.
M15 culture medium: To 500 ml of knockout MEM, addition of 90 ml of ES-cult serum, 6 ml of penicillin streptomycin glutamate, 6 ml of 100 x BME ( to make the 100x stock , add 37 µl of β-ME to 50 ml of PBS) and 60 µl of ESGRO (LIP). Storage should be done for maximum of two months at 4 °C.
M151 x freezing medium: for 100ml of M15 freezing medium, combination of 50 ml of M15 medium, 40 ml of ES- cult serum and 10 ml of DMSO. Storage should be done at 4 °C of maximum of 2-3 months.
Feeder cells medium: To 500 ml of knock out MEM, add 60 ml of ES-cult serum, 6 ml of penicillin- streptomycin –glitamate and 6 ml of 100 x β-ME. Storage should be done at 4 °C for 2-3 months.
Principle:
- Cloning of cDNA of P2X1 and ADRA1A gene in the pVAL vector.
- Cloning of 10, 15 or more different shRNA into pVAL gene of interest pVAL(P2X1 and ADRA1A genes)
- Transfection of NIH3T3 cells with resulting validation vectors.
- Quantification of target gene knockdown by PCR or q RTPCR relative to control cells
Available pVAL platforms:(Sirion biotech)
|
Vactor system |
Promoter |
Transgene |
Pplin |
Strand moduls |
|
pVAl- U6 |
U6 |
shRNA |
Constituted |
10, 15 0r more shRNA |
Procedure: (Lukas E D., et al., 2012)
Designing:Identification of mRNA requence to be targeted has to be recognized from the NCBI database. Design the mIR3097-mer PCR templates for the PCR cloning procedure.
shRNA cloning:
Preparation of PCR templates
Cloning of siRNA by PCR.
PCR cloning:
TABLE 1:Master mix for PCR can be prepared by using pfx platinum polymerase in DNA clean area such as PCR hood. (Lukas E D., te al., 2012)
|
Ingredients |
Amount per well (µl) |
Final |
|
H2O |
32.49 |
|
|
PCR buffer |
4.99 |
1X |
|
MgSO4 (49.99 mM) |
0.99 |
0.99mM |
|
DNTP (2.49 nM each) |
4.99 |
0.24 mM |
|
5’ miRXho (9.99µM) |
1.99 |
0.399µM |
|
3’ miRECO |
1.99 |
0.399µM |
|
pfX platinum polymerase |
0.499 |
1.24 µM |
TABLE 2:Addition of template oligonucleotide (0.02 µg/µl) and run the PCR cycling. (Lukas E D., te al., 2012)
|
Cycle integer |
Denaturation |
Annealing |
Extendation |
|
1 |
93 °C for 4.00 |
|
|
|
2.34 |
93 °C for 0:14 |
54 °C for 0:29 |
67 °C for 0:24 |
|
35 |
-- |
- |
69 °C for 6:00 |
Digestion can be done at 37 °C for 3-4 hrs.
Table 3:Restriction digest (Lukas E D., te al., 2012)
|
constituent |
Concentration per well (µl) |
Final |
|
Purified PCR product |
29.99 |
|
|
ECORI buffer 10X |
4.99 |
1X |
|
BSA(10 mg/ml)100X |
0.49 |
0.1mg/ml |
|
Xhol(19999 U/ml) |
0.99 |
20 U |
|
ECORI (19999 U/ml) |
0.99 |
20 U |
|
DNA free H2O |
12.49 |
- |
Cloning of XhoI/EcoRIsubcloning of shRNAs
Table 4:Digestion of the recipient miR30-based vector(usually pLMP) XhoI/ECORI should be done at 37 °C for 4 hrs.(Lukas E D., te al., 2012)
|
constituent |
Concentration (µl) |
Final |
|
ECO RI buffer |
4.99 |
1X |
|
BSA (10 mg/ml) |
0.49 |
0.099 mg/ml |
|
PLMP (0.99 µg/ml) |
4.99 |
4.99 µg |
|
ECO RI (19999 U/ml) |
1.49 |
29 U |
|
Xhol (19999 U/ml) |
1.49 |
29 U |
|
DNA free H2O |
30.49 |
|
Heat-inactivation of enzymes can take place at 70 °C for 15 minutes.
Addition of 10 U of CIP and incubation of around 1 hr at 37 °C.
Purification of vector backbone can be done with the help of Qiagen PCR purification kit.
TABLE 5:Ligation of 4 nm of digested 110 bp PCR product with 100ng of XhoI/ECORI- digetedpLMP (molecular ratio- 3:1), this mixture should be kept at 15 °C (most efficient).(Lukas E D., te al., 2012)
|
Constituent |
Concentration per well |
Final |
|
Ligation buffer 10X |
0.99 µl |
1X |
|
Xhol/ECORI digested vector |
99.99 µg |
99.99 ng |
|
Xhol/ECORI digested PCR product |
0.99 µl |
3.99 ng |
|
NEB DNA ligase (1999999 U/ml) |
0.49 U/ml |
1000 U |
|
DNase free H2O |
Up to 9.99 µl |
|
A vector only control ligation (no PCR product) is incubated to assess the background colony number after the transformation.
Each ligation reaction must be combined with competent XL10-GOLD bacteria in 1:20 (ligation/bacteria ratio) on ice.
Incubation of the tubes should be done on ice for 10-29 min and then the transformation is required (heat shock or electroporation). It is dependent on the competency of the cells. The individual shRNA cloning and subcloning, the ligation efficiency is always higher for such kind of procedure. Xl-GOLD bacteria can be prepared by using calcium chloride and then transformation of these bacteria can be done by heat shocking at 42 °C for 45 Seconds.
Testing of knockdown : virus production and collection
Placement of plat E cells at 2 × 106cells per 6 cm plate
If the cells are 90 % confluent (usually 6-8 hr later) then transfection of these cells with 5 µg of every miR30 vector with calcium phosphate precipitation agent.
After the tranfection has been done at 18 hrs rinse the cells twise with 4 ml of PBS and then replace it with 25 ml of viral collection medium (DMEM) with 10% vol/vol calf serum.
Collection of viral supernatant 30, 48, and 54 hrs after the tranfection procedure.
After the viral collection has been done, pour supernatant and the debris can be removed by the use of 0.45 µm syringe.
Objective cell transduction and selection:
For every shRNA to be analysed (including the control shRNA), placement of 75,000 NIH cells per well in three wells of six -well plate, 24 hrs before the transduction. For nontransduced control, one additional well used for flow cytometry and antibiotic selection.
To the 2 ml of cell culture medium, addition of 2 µl of polybrene (4mg/ml) and 200 µl, 40 µl or 10 µl of viral supernatant for the generation of working viral dilutions of – 1/10, 1/50 and 1/200.
Incubation of the cells can be done with virus at 37 °C for 24 hrs.
The virus-containing medium should be discarded and recovery of cells must be done in 2 ml of normal culture medium for 24 hrs.
After the tripsinization of cells, the analysis of every transduced can be done by using florescence flow cytometry. And then the percentage of the tranduced cells can be calculated.
For every shRNA, selection of transduced population can be done for GFP positive cells which exhibits the count of 5-20%.
Confirmation of selection of retrovirus can be done by flow cytometry for the expectation results greater than 99% GFP positive cells.
Analysis of measurement of protein knockdown can be analysed by western blot analysis.
Ethical issues
The animals should be housed and identified according to the ISAE guidlines.
The choice of animals and the handling of animals should be according to the section “standard practices” mentiones under ISAE guidelines.
The field of experiment and procedure like euthanasia and cervical dislocation should be done according to ISAE guidelines.
Results
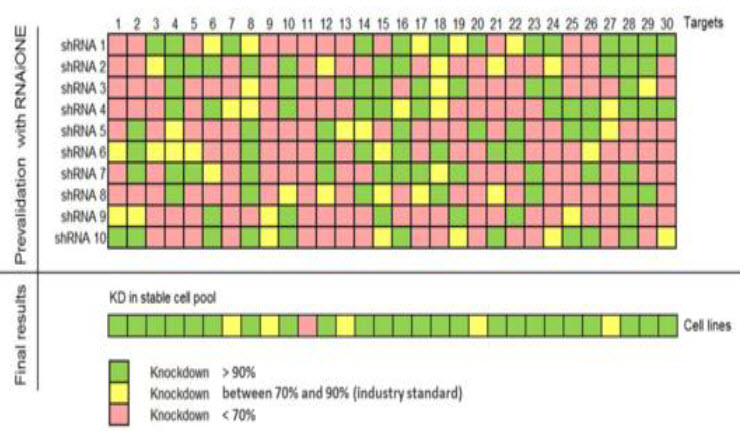
(Sirion biotech)
Chart 1: Presentation of data by SIRION biotech has managed to identify shRNAs with knockdown of about 90% significance interval. (Sirion biotech)
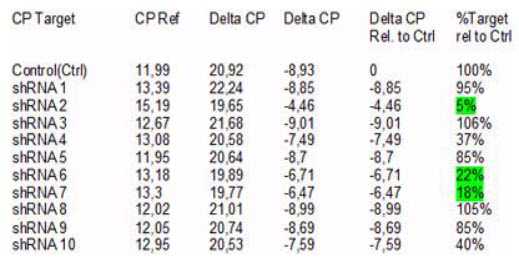
(Sirion biotech)
Table 6: Analysis of sequence check for knockdown: it can be shown that only 2 shRNA achieved a knockdown superior to 90% significance interval. (SIRION biotech)
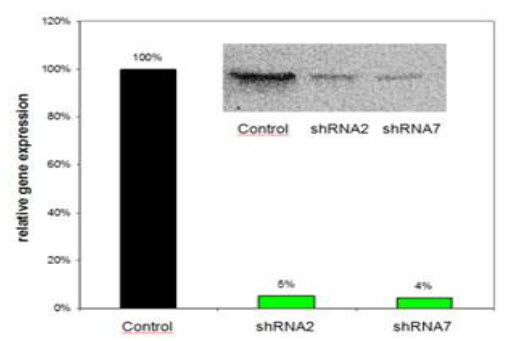
(Sirion biotech)
Graph 1: Results for relative quantification of target gene expression of stable cell lines by western blot and qRT-PCR, NT-shRNA(control) had no effect on gene expression.
By use of rhRNA with collaboration with rhRNAhir technique can produne the transgenic mice.
The transgenic mice can produce the collal targeting contain common transgenic(specific sequences the use of a generic genotyping PCR to recognise any integrated transgene at the locus centre.
By inhibiting the genes for the proteins male can become 100% infertile.
The cell lines that exhibits the 90% knockdown has the more significance on transgenic mice which have the 100% infertility for male mice specimens.
Outcomes & Conclusion
The two proteins α1A receptor and P2X1 receptor has been responsible for the male fertility. By knocking down the genes responsible for the expression of these proteins male animals can become infertile.
The simple PCR based approach can be used for the production of different shRNAmir transgenic mice. The two prtiens alpha 1A-adrenoceptor and P2X1-purineceptor are both receptor molecules on which other agents cut to trigger biological process.
The knockdown rhRNAmir mice have the same sex drive or normal sexual activity. The mice may appear normal and may have no side-effects. Trials are going on for human beings besides the function for other organs in body should be considered for evaluation.
References
* Gilbert LA, “CSISPR-mediated modular RNA-guided regulation of transcription in eukaryotes” Cell, 2013, 152 442-451.
* Kamath R.S., “RNA therapeutics principles prospects and challenges” Advanced drug delievery Review, 2007, 59(2-3) 75-86.
* Lukas E D., Prem K.P., Johannes Z., Christ F., Katherine, Mcjulin, Corneliws, Meithing, Youngkyv P., Ross A.D., Gregoly J.H. and Scott W.L., “A pipeline for the generation of shRNA transgenic moce” Nat Protoc, 2012, 7(2), 374-393.
* Summerton J, “MorpholinoSiRNA, and S’-DNA compared: Impact of structure and mechanism of action on off-target effects and sequence specificity” Med Chem, 2007, 7(7), 651-660.
* Summerton J, Morpholio Antisense oligomers: The case for an RNase – H dependent structure type Biochemica et Biophysica Acta, 1999, 1489(1), 141-58.
* Sun N., “Transcription activator like effector nucleases(TALENS) : a highly efficient and versetile tool for genome editing” Bitotechnology and bioengineering, 2013, 110 1811-1821.
|
PharmaTutor (ISSN: 2347 - 7881) Volume 2, Issue 4 Received On: 11/02/2014; Accepted On: 19/02/2014; Published On: 01/04/2014 How to cite this article: EM Patelia, E Spiking, Knockdown (Micro-RNA Inhibiting Technique) Technique for Evaluation of Alpha-1A Adrenergic Receptor and P2X1 Receptor Functions, PharmaTutor, 2014, 2(4), 119- 125 |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
FIND OUT MORE ARTICLES AT OUR DATABASE








.png)

