

About Authors:
Tarun Garg, Ajay Bilandi,
Seth G.L.Bihani S.D.college of technical education,
Sri Ganganagar
ABSTRACT
The aim of this review is to improve the solubilization and bioavailability of poorly soluble drugs by using various approaches like physical, chemical and others modifications or techniques. The solubility of a solute is the maximum quantity of solute that can dissolve in a certain quantity of solvent or quantity of solution at a specified temperature. Solubility is one of the important parameter to achieve desired concentration of drug in systemic circulation for pharmacological response to be shown. Drug efficacy can be severely limited by poor aqueous solubility and some drugs also show side effects due to their poor solubility. There are many techniques which are used to enhance the aqueous solubility. The ability to increase aqueous solubility can thus be a valuable aid to increasing efficiency and/or reducing side effects for certain drugs. This is true for parenterally, topically and orally administered solutions. Physical modifications techniques like media milling/ nanocrystal technology, cryogenic technology, supercritical fluid process, modification of the crystal habit,complexation, micellar technologies, chemical modifications, other techniques like co-crystallization, cosolvency, hydrotrophy are used for increase the solubility of very soluble drugs like dolargin, loperamide, tubocurarine, doxorubicin, ibuprofen, griseofulvin, diazepam, naproxen, carbamazepine, nifedipine, phytosterol etc.
Refernce Id: PHARMATUTOR-ART-1153
INTRODUCTION
Solubilization of poorly soluble drugs is a frequently encountered challenge in screening studies of new chemical entities as well as in formulation design and development [1,2]. A number of methodologies can be adapted to improve solubilization of poor water soluble drug and further to improve its bioavailability. Orally administered drugs completely absorb only when they show fair solubility in gastric medium and such drugs shows good bioavailability. Bioavailability depends on several factors, drug solubility in an aqueous environment and drug permeability through lipophilic membranes being the important ones [3]. The techniques generally employed for solubilization of drug includes micronization, chemical modification, pH adjustment, solid dispersion, complexation, co?solvency, micellar solubilization, hydrotropy etc. Actually, only solubilized drug molecules can be absorbed by the cellular membranes to subsequently reach the site of drug action (vascular system for instance). Any drug to be absorbed must be present in the form of an aqueous solution at the site of absorption [4-7]. As Solubility & permeability is the deciding factor for the in-vivo absorption of the drug, these can be altered or modified by enhancement techniques like [8]. Poorly soluble compounds belongs to class II of BCS, also present many in vitro formulation obstacles, such as severely limited choices of delivery technologies and increasingly complex dissolution testing with limited or poor correlation to the in vivo absorption. Recently more than 40% NCEs (new chemical entities) developed in Pharmaceutical Industry are practically insoluble in water. These poorly water soluble drugs are allied with slow drug absorption leading to inadequate and variable bioavailability and gastrointestinal mucosal toxicity [9]. Therefore, the improvement of drug solubility thereby its oral bio-availability remains one of most challenging aspects of drug development process especially for oral drug delivery system. These in vivo and in vitrocharacteristics and the difficulties in achieving predictable and reproducible in vivo/in vitro correlations are often sufficiently difficult to develop formulation on many newly synthesized compounds due to solubility issues [10,11]. Although pharmaceutical companies have been able to overcome difficulties with very slightly soluble drugs, those with aqueous solubility of less than 0.1 mg/ml present some unique challenges. This review thus begins with discussion regarding the traditional approaches to drug solubilisation include Ph adjustment, cosolvency and particle size reduction. While microemulsion and self-emulsifying systems are novel approaches.These drugs are particularly good candidates for advanced solubilization technologies developed by companies specializing in drug delivery. Traditional approaches to drug solubilisation include particle size reduction, pH adjustment and addition of surfactants and cosolvents, while microemulsion and self-emulsifying systems are novel approaches. There are numerous approaches available and reported in literature to enhance the solubility of poorly water soluble drug. The techniques are chosen on the basis of certain aspects such as properties of drug under consideration, nature of excipients to be selected and nature of intended dosage form. This review is intended to discuss the various traditional and novel techniques for solubility enhancement of hydrophobic drugs for oral pharmaceutical formulation.
PROCESS OF SOLUBILISATION
The process of solubilisation involves the breaking of intermolecular or inter-ionic bonds in the solute, the separation of the molecules of the solvent to provide space in the solvent for the solute, interaction between the solvent and the solute molecule or ion [12]. Solubilisation process occurs into three steps. (Fig. 1).
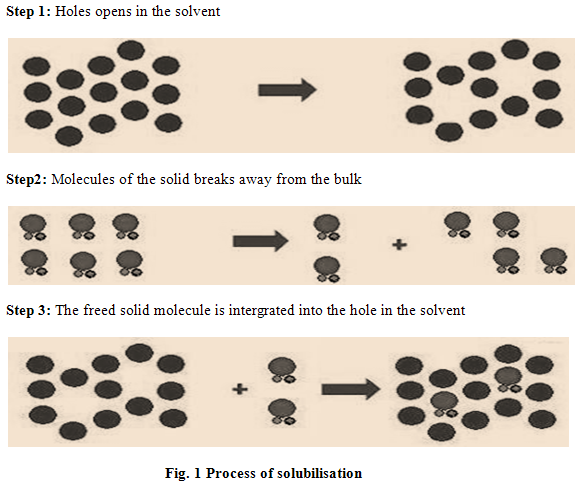
FACTORS AFFECTING SOLUBILITY
The solubility depends on the nature and composition of solvent medium, the physical form of the solid as well as temperature and pressure of system. Consider a lot of factor, which affects the solubility like-
(1) Particle Size
The size of the solid particle influences the solubility because as a particle becomes smaller, the surface area to volume ratio increases of the particle. The larger surface area allows a greater interaction with the solvent. The effect of particle size on solubility can be described
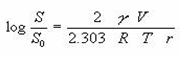
Where, S0 is the solubility of infinitely large particles, S is the solubility of fine particles, V is molar volume, r is the radius of the fine particle and γ is the surface tension of the solid.
(2)Temperature
As the temperature is increased than the solution process absorbs energy and the solubility will be increased but if the solution process releases energy then the solubility will decrease with increasing temperature. A few solid solutes are less soluble in warm solutions. For examples all gases, solubility decreases as the temperature of the solution increases.
(3) Pressure
For solids and liquid solutes, changes in pressure have practically no effect on solubility but for gaseous solutes, an increase in pressure, increases solubility and a decrease in pressure, decrease the solubility.
(4) Nature of the solute and solvent
Only 1 gram of lead (II) chloride can be dissolved in 100 grams of water at room temperature while 200 grams of zinc chloride can be dissolved. The great difference in the solubility’s of these two substances is the result of differences in their natures.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
(5) Molecular size
The solubility of the substance is decreased when molecules have higher molecular weight and higher molecular size because larger molecules are more difficult to surround with solvent molecules in order to solvate the substance. In the case of organic compounds the amount of carbon branching will increase the solubility since more branching will reduce the size (or volume) of the molecule and make it easier to solvate the molecules with solvent.
(6) Polarity
Polarity of the solute and solvent molecules will affect the solubility. Generally like dissolves like means non-polar solute molecules will dissolve in non-polar solvents and polar solute molecules will dissolve in polar solvents. The polar solute molecules have a positive and a negative end to the molecule. If the solvent molecule is also polar then positive ends of solvent molecules will attract negative ends of solute molecules. This is a type of intermolecular force known as dipole-dipole interaction. The other forces called london dispersion forces where the positive nuclei of the atoms of the solute molecule will attract the negative electrons of the atoms of a solvent molecule. This gives the non-polar solvent a chance to solvate the solute molecules.
(7) Polymorphs
Polymorphs can vary in melting point. Since the melting point of the solid is related to solubility, so polymorphs will have different solubility’s. Generally the range of solubility differences between different polymorphs is only 2-3 folds due to relatively small differences in free energy.
(8) Rate of solution
The rate of solution is a measure of how fast substances dissolve in solvents. A various factors affecting rate of solution like-
(a) Size of the particles
Breaking a solute into smaller pieces increases its surface area, when the total surface area of the solute particles is increased; the solute dissolves more rapidly because the action takes place only at the surface of each particle and hence increases its rate of solution.
(b)Temperature For liquids and solid solutes, increasing the temperature not only increases the amount of solute that will dissolve but also increases the rate at which the solute will dissolve. For the gases, reverse is true.
(c) Amount of solute already dissolved
When there is little solute already in solution, dissolution takes place relatively rapidly. As the solution approaches the point where no solute can be dissolved, dissolution takes place more slowly.
(d) Stirring
With liquid and solid solutes, stirring brings fresh portions of the solvent in contact with the solute, thereby increasing the rate of solution [13].
TECHNIQUES OF SOLUBILITY AND BIOAVAILABILITY ENHANCEMENT
There are various techniques available to improve the solubility of poorly soluble drugs. Some of the approaches to improve the solubility are:
(1) pH adjustment
It is well documented that the influence of the changes in pH within the gastrointestinal tract upon the bioavailability of pharmaceuticals. The absorption of drug is largely dependent upon diffusion, which varies with pH of the individual regions within the gastrointestinal tract, the pKa of the drug and permeability, which are not only moderated by the surface area of the region in which it is released, but also the regional pH effects upon drug ionization. By applying a pH change, poorly water soluble drugs with parts of the molecule that can be protonated (base) or deprotonated (acid) may potentially be dissolved in water. While the importance of critical parameters like salt selection and pH adjustment has been stressed on pre-formulation, the use of pH-altering excipients within drug delivery systems is also of significant utility. pH adjustment can in principle be used for both oral and parenteral administration. Because blood is a strong buffer, upon intravenous administration the poorly soluble drug may be precipitate with pH between 7.2 – 7.4. To assess the suitability of the approach, the buffer capacity and tolerability of the selected pH are important to consider. In the stomach the pH is around 1 to 2 and in the duodenum the pH is between 5-7.5, so upon oral administration the degree of solubility is also likely be influenced as the drug passes through the intestines. Solubilised excipients that increase environmental pH within a dosage form (tablet or capsule), to a range higher than pKa of weakly-acidic drugs increases the solubility of that drug, those excipients which act as alkalising agents may increase the solubility of weakly basic drugs [14,15]. After pH adjustment, ionisable compounds (may be acids or bases or zwitterionic) are stable and soluble. It can also be applied to crystalline as well as lipophilic poorly soluble compounds [16-18]. Bioavailability can be increased, if the precipitation upon dilution is fine or amorphous, due to an increased concentration gradient and enhanced surface area for dissolution. In situations where the drug precipitates into poorly soluble particles that require dissolution and do not rapidly redissolve, bioavailability may not be sufficiently increased. The solubility of the poorly soluble drug is increased compared to water alone, so if compounds can permeate through the epithelium orally, the fraction of orally absorbed drug may be increased. pH adjustment is also frequently combined with co-solvents to further increase the solubility of the poorly soluble drug. This approach is used frequently in survey as pre-clinically pH adjustment is a good technique to assess the efficacy of poorly soluble drugs due to its universality and relative simplicity. Advantage of this technique like simple to formulate and analyse, simple to produce and fast track, uses small quantities of compound and amenable to high throughput evaluations. There are some serious disadvantages like risk for precipitation upon dilution with aqueous media having a pH at which the compound is less soluble. Intravenously this may lead to emboli, orally it may cause variability and toxicity (local and systemic) related with the use of a non physiological and extreme pHs. However, if precipitation of the poorly soluble drug occurs uncontrollably after contact with a pH at which the drug is much less soluble (oral as well as parenteral), the interpretation of the results may be misleading. Another problem with this adjustment is that selected pH may accelerate hydrolysis or catalyze other degradation mechanisms because a dissolved drug in an aqueous environment is frequently less stable chemically compared to crystalline solid [19-22].As with all solubilized and dissolved systems, a dissolved drug in an aqueous environment is frequently less stable chemically compared to formulations crystalline solid. Commercial products using pH adjustment :Phenytoin Injection (Epanutin® ready mixed, Pfizer) 50mg/ml with propylene glycol 40% and ethanol 10% (1.1 mmol Na+ per 5 ml ampoule) is an example of a pH adjusted formulation containing co-solvents.
(2)Microemulsion
A micro emulsion is an optically clear pre-concentrate, isotropic, thermodynamicallystable transparent (or translucent) system, containing a mixture of oil, hydrophilic surfactant and hydrophilic solvent which dissolves a poorly water soluble drug. Upon contact with water, the formulations spontaneously disperse (or ‘self emulsifies’) to form a very clear emulsion of exceedingly small and uniform oil droplets containing the solubilised poorly soluble drug.Microemulsions have been employed to increase the solubility of many drugs that are practically insoluble in water, along with incorporation of proteins for oral, parenteral, as well as percutaneous/transdermal use [23,24].These homogeneous systems, which can be prepared over a wide range of surfactant concentration and oil to water ratio, are all fluids of low viscosity.Surfactants, surfactant mixtures and cosurfactants in microemulsions play animportant role in improving the solubility of drugs formulated as microemulsions.An anhydrous system of microemulsions is that self microemulsifying drug delivery system (SMEDDS) or microemulsion pre-concentrate. It is composed of oil, surfactant and cosurfactant and has the ability to form o/w microemulsion when dispersed in aqueous phase under gentle agitation. The agitation required for the self-emulsification comes from stomach and intestinal motility [25-27]. The surfactant like polyoxyethylene surfactants e.g. Brij 35 or sugar esters like sorbitan monooleate (Span 80), cationic, or anionic like alkyltrimethylammonium bromide and sodium dodecyl sulphate, or zwitterionic such as phospholipids like lecithin (phosphatidylcholine) commercially available from soybean and eggs, can be non-ionic. Lecithinis very popular because it exhibits excellent biocompatibility. Due to the liquid nature of the product, most self-emulsifying systems are limited to administration in lipid-filled soft or hard-shelled gelatin capsules. Interaction between the capsule shell and the emulsion should be considered so as to prevent the hygroscopic contents from dehydrating or migrating into the capsule shell [28,29]. Combinations of ionic and non-ionic surfactants are also found to be effective. Microemulsion preconcentrates remain optically clear after dilution and usually contain a higher amount of water soluble surfactant and a higher content of a hydrophilic solvent compared to macroemulsion pre-concentrates. Due to the nature of the excipients, these formulations are only administered orally. Solubilization using microemulsion pre-concentrates is suited to poorly soluble lipophilic compounds that have high solubility in the oil and surfactants mixtures. Advantages: The pre-concentrates are relatively easy to manufacture. Well developed microemulsion pre-concentrates are not normally dependent upon digestion for drug release. Therefore, optimal bioavailability and reproducibility can be also being expected without co-administration of food (i.e. the fasted state).The major disadvantage of microemulsions is their high concentration of surfactant /cosurfactant, making them unsuitable for IV administration. Dilution of microemulsions below the critical micelle concentration of the surfactants could cause precipitation of the drug; however, the fine particle size of the resulting precipitate would still enhance absorption .The precipitation tendency of the drug on dilution may be higher due to the dilution effect of the hydrophilic solvent. The tolerability of formulations with high levels of synthetic surfactants may be poor in cases where long term chronic administration is intended. Formulations containing several components become more challenging to validate [30,31]. Microemulsion products:Examples of poorly soluble compounds that use micro-emulsion pre-concentrates are the HIV protease inhibitor tipranavir (Aptivus® capsules, Boehringer Ingelheim GmBH) and the category defining immunosuppressant cyclosporine A, USP modified (Neoral® capsules, Novartis AG) [32].
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
(3) Self-emulsifying drug delivery systems
Self-emulsifying or self-micro emulsifyingsystems use the concept of in situformation of emulsion in thegastrointestinal tract. The mixture of oil,surfactant, cosurfactant, one or morehydrophilic solvents and cosolvent forms a transparent isotropic solution that is known as the self-emulsifying drug delivery system (SEDDS) [33], in the absence ofexternal phase (water) and forms fine o/w emulsions or microemulsions spontaneously upon dilution by theaqueous phase in the GIT and is used for improving lipophilic drug dissolution and absorption. The ease of emulsification could be associated with the ease of water penetrating into the various liquids crystalline or gel phases formed on thesurface of the droplet. One of the advantages of SEDDS in relation to scaleupand manufacture is that they form spontaneously upon mixing their components under mild agitation and they are thermodynamically stable. The drawbacks of this system include chemical instabilities of drugs and high surfactant concentrations. The large quantity of surfactant in self-emulsifying formulations(30-60%) irritates GIT. Most self-emulsifying systems are limited to administration in lipid-filled soft or hard-shelled gelatine capsules due to the liquid nature of the product. Interaction between the capsule shell and the emulsion should be considered so as to prevent the hygroscopic contents from dehydrating or migrating into the capsule shell [34].A Neoral® is an example of selfmicroemulsfying drug delivery system (SMEDDS). Depending on the dose level, the relative bioavailability of cyclosporine A administered as Neoral® could be 174- 239% of the bioavailability of cyclosporine A from Sandimmune®, the originally marketed formulation [35]. Emulsion droplet size is a major factor influencing bioavailability of drugs from emulsion formulations, with small droplet radii enhancing the plasma levels of drugs,in part due to direct lymphatic uptake. Since SMEDDS contain high concentration of surfactants, they should be limited to oral applications and may not be advisable for long-term use due to the potential of causing diarrhoea [36].
(4) Manipulation of solid state
From the stability and bioavailability aspects, the crystalline form of a drug is of pharmaceutical importance. Polymorphism (existence of a drug substance in multiple crystalline forms) can cause variations in melting point, density, stability and drug solubility as these properties depend on the escaping tendency of the molecules from a particular crystalline structure. As a rule, for a drug that have the highest order of crystallinity is the most stable form, exists in multiple polymorphic forms, i.e. with the least amount of free energy, and, consequently, possesses the highest melting point and the least solubility. By controlling the crystallisation process, amorphous or metastable forms of drugs possessing high free energy can be forcibly created. They offer the advantage of higher solubility but suffer from stability issues unless stabilisers intended to inhibit crystal growth are incorporated in the formulation [37]. A high profile case involving polymorphism was withdrawal of ritonavir (Norvir®) capsules from the market in 1998 because a less soluble (and consequently less bioavailable) polymorph was identified two years after the product was approved and marketed, causing a decrease in bioavailability of the drug [38]. This incident sensitized the pharmaceutical industry to the critical importance of polymorphism and encouraged the inclusion of polymorph screening as a routine component of preformulation studies.
(5)Particle size reduction
The bioavailability of poorly soluble drugs is often intrinsically related to drug particle size. By reducing particle size, the increased surface area may improve the dissolution properties of the drug to allow a wider range of formulation approaches and delivery technologies [39]. The larger surface area allows a greater interaction with the solvent which cause increase in solubility [40]. Conventional methods of particle size reduction, such as comminution and spray drying, rely upon mechanical stress to disaggregate the active compound. The mechanical forces inherent to comminution, such as milling and grinding, often impart significant amounts of physical stress upon the drug product which may induce degradation. During comminution and spray drying is also a concern when processing thermosensitive or unstable active compounds, the thermal stress which may occur. Also, this traditional methods are often incapable of reducing the particle size of nearly insoluble drugs (<0.1mg/mL) [41]. Nowadays Particle size reduction can be achieved by micronisation and nanosuspension. Each technique utilizes different equipments for reduction of the particle size. In micronization the solubility of drug is often intrinsically related to drug particle size. By reducing the particle size, the increased surface area improves the dissolution properties of the drug. Micronization of drugs is done by milling techniques using jet mill, rotor stator colloid mills etc. Micronization is not suitable for drugs having a high dose number because it does not change the saturation solubility of the drug. These processes were applied to griseofulvin, progesterone, spironolactone and diosmin, fenofibrate. For each drug, micronization improved their digestive absorption, and consequently their bioavailability and clinical efficacy [42]. Nanosuspension is another technique which is sub-micron colloidal dispersion of pure particles of drug, which are stabilised by surfactants. The nanosuspension approach has been employed for drugs including tarazepide, atovaquone, amphotericin B, paclitaxel and bupravaquon.The advantages offered by nanosuspension is increased dissolution rate is due to larger surface area exposed, while absence of Ostwald ripening is due to the uniform and narrow particle size range obtained, which eliminates the concentration gradient factor. Liquid forms can be rapidly developed for early stage testing (pre-clinical) that can be converted into solids for later clinical development. Typically, low excipient to drug ratios is required.Formulations are generally well tolerated provided that strong surfactants are not required for stabilisation. Generally, crystal forms are chemically and physically more stable than amorphous particles. A method to consider for stubborn compounds that defeat previous attempts to increase solubility. Nanosuspensions are produced by homogenization and wet milling process [43]. Recrystallisation of poorly soluble materials using liquid solvents and antisolvents has also been employed successfully to reduce particle size. The reliance upon organic solvents during processing often involves solvent extraction and handling procedures which may significantly increase the complexity of manufacture.Disadvantages: Due to the high surface charge on discrete small particles, there is a strong tendency for particle agglomeration. Developing a solid dosage form with a high pay load without encouraging agglomeration may be technically challenging. Technically, development of sterile intravenous formulations is even more challenging.
Ball milled products: This process is widely used in nonpharmaceutical applications particularly in cosmetics to obtain ultra fine particles for sun block. Examples of pharmaceutical products include rapamycin (Rapamune®, 1 mg and 2 mg tablets, Wyeth).
(6)Supercritical fluid (SCF) process
Another novel nanosizing and solubilisation technology whose application has increased in recent years is particle size reduction via supercritical fluid (SCF) processes. The number of applications and technologies involving supercritical fluids has also grown explosively. It has been known for more than a century that supercritical fluids (SCFs) can dissolve nonvolatile solvents, with the critical point of carbon dioxide, the most widely used supercritical fluid.Supercritical fluids are fluids whose temperature and pressure are greater than its critical temperature (Tc) and critical pressure (Tp), allowing it to assume the properties of both a liquid and a gas.It is safe, environmentally friendly, and economical. The low operating conditions (temperature and pressure) make SCFs attractive for pharmaceutical research.At near-critical temperatures, SCFs are high compressible, allowing moderate changes in pressure to greatly alter the density and mass transport characteristics of a fluid that largely determine its solvent power [44]. A SCF exists as a single phase above its critical temperature (Tc) and pressure (Pc). SCFs have properties useful to product processing because they are intermediate between those of pure liquid and gas (i.e., liquid-like density, gas-like compressibility and viscosity and higher diffusivity than liquids).At near-critical temperatures, SCFs are high compressible, allowing moderate changes in pressure to greatly alter the density and mass transport characteristics of a fluid that largely determine its solvent power.Moreover, the density, transport properties (such as viscosity and diffusivity), and other physical properties (such as dielectric constant and polarity) vary considerably with small changes in operating temperature, pressure, or both around the critical points [45]. Once the drug particles are solubilised within SCF, they may be recrystallised at greatly reduced particle sizes. The flexibility and precision offered by SCF processes allows micronisation of drug particles within narrow ranges of particle size, often to sub-micron levels.The flexibility and precision offered by SCF processes allows micronisation of drug particles within narrow ranges of particle size, often to sub-micron levels. Hence, it is possible to fine-tune a unique combination of properties necessary for a desired application. These unique processing capabilities of SCFs, long recognized and applied in the food industry, have recently been adapted to pharmaceutical applications. Current SCF processes have demonstrated the ability to create nanoparticulate suspensions of particles 5- 2,000nm in diameter.Several pharmaceutical companies, such as Nektar Therapeutics and Lavipharm, are specialising in particle engineering via SCF technologies for particle size reduction and solubility enhancement [46]. Commonly used supercritical solvents include carbon dioxide, nitrous oxide, ethylene, propylene, propane, n-pentane, ethanol, ammonia, and water. Several methods of SCF processing have been developed to address individual aspects of these shortcomings, such as precipitation with compressed antisolvents process (PCA), solution enhanceddispersion by SCF (SEDS), supercritical antisolvents processes (SAS) and aerosol supercritical extraction system (ASES) [47].
(7)Inclusion complexes/ complexation
Lipophilic drug-cyclodextrin complexes, commonly known as inclusion complexes, can be formed simply by adding the drug and excipient together, resulting in enhanced drug solubilization. Among all the solubility enhancement techniques inclusssion complex formation technique has been employed more precisely to improve the aqueous solubility, dissolution rate, and bioavailability of poorly water soluble drugs.Cyclodextrins (CD) are a group of structurally-related cyclic oligosaccharides that have a polar cavity and hydrophilic external surface. Inclusion complexes are formed by the insertion of the nonpolar molecule or the nonpolar region of one molecule (known as guest) into the cavity of another molecule or group of molecules (known as host). The most commonly used host molecules are cyclodextrins. Cyclodextrins consisting of 6, 7 and 8 D glucopyranosyl units connected to α -1, 4 glycosidic linkages are known as α, β, γ, cyclodextrins, respectively [48]. Derivatives of β-cyclodextrin with increased water solubility (e.g. hydroxypropyl-β-cyclodextrin HP-β-CD) are most commonly used in pharmaceutical formulation. Cyclodextrins consist of glucose monomers arranged in a donut shape ring [49].Hydrophilic cyclodextrins are nontoxic in normal doses while lipophilic ones may be toxic; hence, methyl, hydroxypropyl, sulfoalkylated and sulfated derivatives of natural cyclodextrins that possess improved aqueous solubility are preferred for pharmaceutical use. The ring has a hydrophilic exterior and lipophilic core in which appropriately sized organic molecules can form noncovalent inclusion complexes resulting in increased aqueous solubility and chemical stability [50].The forces driving complexation were attributed to (i) the exclusion of high energy water from the cavity, (ii) the release of ring strain particularly in the case of α -CD, (iii) Vander walls interactions, and (iv) hydrogen and hydrophobic bindings [51]. Cyclodextrin complexes have been shown to increase the stability, wettability and dissolution of the lipophilic insect repellent N, N-diethyl-m-toluamide (DEET) and the stability and photostability of sunscreens. Cyclodextrins are large molecules, with molecular weights greater than 1000Da, therefore it would be expected that they would not readily permeate the skin. Complexation with cyclodextrins has been variously reported to both increaseand decrease skin penetration [52].Solublization by complexation is achieved through specific interaction rather than changes in the bulk solvent properties as in other solublizing system such as cosolvents, emulsion and pH adjustments. In a recent review of the available data, Loftsson and Masson concluded that the effect on skin penetration may be related to cyclodextrin concentration, with reduced flux generally observed at relatively high cyclodextrin concentrations, whilst low cyclodextrin concentrations resulting in increased flux [53]. As flux is proportional to the free drug concentration, where the cyclodextrin concentration is sufficient to complex only the drug which is in excess of its solubility, an increase in flux might be expected. The dissociation is very rapid, quantitative and therefore predictable. Another significant advantage of complexation technique is that some commonly used complexing agents such as hydroxy propyl beta cyclodextrin and sulfobutyl beta cyclodextrin are less toxic compared to other solublizing agents such as surfactant and cosolvents. However, at higher cyclodextrin concentrations, the excess cyclodextrin would be expected to complex free drug and hence reduce flux. Skin penetration enhancement has also been attributed to extraction of stratum corneum lipids by cyclodextrins. Given that most experiments that have reported cyclodextrin mediated flux enhancement have used rodent model membranes in which lipid extraction is considerably easier than human skin [54], the penetration enhancement of cyclodextrin complexation may be an overestimate.Since most complexes formed is 1:1 complexes of the AL type, the dilution of complexes will not result in solution which is supersaturated with respect to substrate. This can be important for very insoluble compounds that may precipitate upon injection when solublized by other system such as cosolvents. Shaker and colleagues recently concluded that complexation with HP- β-CD had no effect on the flux of cortisone through hairless mouse skin by either of the proposed mechanisms [55]. The solubility enhancement application, CDs can also be used as membrane permeability enhancer and stabilizing agents. The permeability through biological membrane is enhanced by the presence of cyclodextrins. CDs can also be used as nasal permeation enhancers acting by interaction with nasal epithelium by modifying tight junction & lipid and protein content of the membrane, which enhances the permeation of the membrane. CDs can also be utilized as permeation enhancer in pulmonary drug delivery systems. Rifampicin is a so- called concentration-dependent antibiotic, the rate and extent of bacterial kill is related to the attainment of high maximum concentration relative to the minimal inhibitory concentration. The rifampicin-CD inclusion compound can improve the lung transport of drug when nebulized with compatible pulmonary deposition and achieve required concentration of drug in broncho-alveolar epithelium lining-fluid when administered as aerosolized solution.The enzymatic degradation of starch by cyclodextrin-glycosyltransferase (CGT) produces cyclic oligomers, Cyclodextrins. Cyclodextrins are non-reducing, crystalline, water soluble, cyclic, oligosaccharides. Solubility and oral bioavailability of Glipizide53, Rofecoxib54, Piroxicam55 and Carvedilol56 can be improved by using cyclodextrins inclusion complex. Solublization by complexation is achieved through specific interaction rather than changes in the bulk solvent properties as in other solublizing system such as cosolvents, emulsion and pH adjustments. The dissociation is very rapid, quantitative and therefore predictable. Masson [56] reported about the permeation enhancement property of poorly water soluble drugs in presence of the CDs. These acts as permeation enhancers by carrying the drug through the aqueous barrier which exists before the lipophilic surface of biological membranes. This can also be achieved through the double characteristics of the CDs, thus present character much lipophilic as hydrophilic. Despite all the attractive advantage of complexation, there are disadvantages. First of all the compound has to be able to form complexes with selected ligand. For compounds with very limited solubility to start with, solubility enhancement can be very limited. The second limitation is the complexes of Ap type, dilution of system may still result in precipitation. This is also true for solublization via combined technique such as complexation with pH adjustment. Lastly the potential toxicity issue, regulatory and quality control issue related to presence of ligand may add complication and cost to the development process [57]. Despite all the attractive advantage of complexation, there are disadvantages. First of all the compound has to be able to form complexes with selected ligand. For compounds with very limited solubility to start with, solubility enhancement can be very limited. The second limitation is the complexes of Ap type, dilution of system may still result in precipitation. This is also true for solublization via combined technique such as complexation with pH adjustment. Lastly the potential toxicity issue, regulatory and quality control issue related to presence of ligand may add complication and cost to the development process [58]. There are various technologies (Table 1) adapted to prepare the inclusion complexes of poorly of poorly water soluble drugs with cyclodextrins.
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
Table 1. Various technologies to prepare inclusion complexes of poorly of poorly water soluble drugs with cyclodextrins
|
S.no. |
Name of techniques |
Detail of techniques |
|
1. |
Kneading method |
This method is based on impregnating the CDs with little amount of water or hydroalcoholic solutions to converted into a paste. The drug is then added to the above paste and kneaded for a specified time. The kneaded mixture is then dried and passed through sieve if required [59]. Parik et al. [60] have reported the dissolution enhancement of nimesulide using complexation method. |
|
2. |
Lyophilization/ Freeze drying technique |
In this technique, the solvent system from the solution is eliminated through a primary freezing and subsequent drying of the solution containing both drug & CD at reduced pressure. Thermolabile substances can be successfully made into complex form by this method. The limitations of this technique are long time process and yield poor flowing powdered product. Lyophilization/ freeze drying technique are considered as an alternative to solvent evaporation and involve molecular mixing of drug and carrier in a common solvent [61]. |
|
3 |
Microwave irradiation method |
This technique involves the microwave irradiation reaction between drug and complexing agent using a microwave oven. The drug and CD in definite molar ratio are dissolved in a mixture of water and organic solvent in a specified proportion into a round bottom flask. The mixture is reacted for short time of about one to two minutes at 60 °C in the microwave oven. After the reaction completes, adequate amount of solvent mixture is added to the above reaction mixture to remove the residual, uncomplexed free drug and CD. The precipitate so obtained is separated using whatman filter paper, and dried in vaccum oven at 40 °C for 48 hrs. |
|
4 |
Supercritical Antisolvent technique |
Supercritical carbon dioxide is suggested as a new complexation medium due to its properties of improved mass transfer and increased solvating power. This method constitutes one of the most innovators methods to prepare the inclusion complex of drug with CD in solid state. This is a non-toxic method as it is not utilizing any organic solvent, fast process, maintenance cost is low with promising results, but it requires a quite high initial cost [62]. |
(8)Co-solvency
The solubility of a poorly water soluble drug can be increased frequently by the addition of a water miscible solvent in which the drug has good solubility known as cosolvents [63]. Co-solvents are mixtures of water and one or more water miscible solvents used to create a solution with enhanced solubility for poorly soluble compounds. Historically, this is one of the most widely used techniques because it is simple to produce and evaluate Cosolvency has been utilised in different formulations including solids and liquids. Examples of solvents used in co-solvent mixtures are PEG 300, propylene glycol or ethanol. Various concentrations (5-40%) of the solid binary systems with polyethylene glycol 6000 were employed to increase solubility and dissolution of meloxicam. Co-solvent formulations of poorly soluble drugs can be administered orally and parenterally. Parenteral formulations may require the addition of water or a dilution step with an aqueous media to lower the solvent concentration prior to administration. Cosolvencytechniques have also found use in spray freezing of liquid like in danazol withpolyvinyl alcohol, poloxamer 407, and polyvinylpyrrolidone K-15 in a micronized powder formulation. The pharmaceutical form is always liquid. Poorly soluble compounds which are lipophilic or highly crystalline that have a high solubility in the solvent mixture may be suited to a co-solvent approach. Co- Solvents can increase the solubility of poorly soluble compounds several thousand times compared to the aqueous solubility of the drug alone. Very high drug concentrations of poorly solublecompounds can be dissolved compared to other solubilization approaches. Though cosolvency has been highly utilised in the design of many different formulations, it has found its main use in parenteral dosage forms because of the irritating effects of most surfactants and the low toxicity of many cosolvents, and because of the relatively greater ability of cosolvents to solubilise nonpolar drugs. The most frequently used low toxicity cosolvents for parenteral use are propylene glycol, ethanol, glycerin, and polyethylene glycol [64,65]. The use of co-solvents is a highly effective technique to enhance the solubility of poorlysoluble drugs [66]. Seedher and Bhatia (2003) investigated that the aqueous solubility of celecoxib, rofecoxib and nimesulide could be enhanced significantly by using ethanol as the second solvent and PEG-400-ethanol had highest solubilization potentiality among the mixed solvent systems. However, the bioavailability may not be dramatically increased because the poorly soluble drug will typically uncontrollably crash out upon dilution into a crystalline or amorphous precipitate. In this case, dissolution of this precipitate is required for oral absorption. Co-solvents may be combined with other solubilization techniques and pH adjustment to further increase solubility of poorly soluble compounds. Dimethylsulfoxide (DMSO) and dimethylacetoamide (DMA) have beenwidely used as cosolvents because of their large solubilization capacity for poorly soluble drugs and their relatively low toxicity [51-54].Advantages: Simple and rapid to formulate and produce. Disadvantages: As with all excipients, the toxicity and tolerability related with the level of solvent administered has to be considered.Uncontrolled precipitation occurs upon dilution with aqueous media. The precipitates may be amorphous or crystalline and can vary in size. Many of the insoluble compounds Phares works with are unsuited to co-solvents alone, particularly for intravenous administration. This is because the drugs are extremely insoluble in water and do not readily redissolve after precipitation from the co-solvent mixture. In these situations, there is a potential risk for embolism and local adverse effects at the injection site.As with all solubilized forms, the chemical stability of the insoluble drug is worse than in a crystalline state. Co-solvent products:Nimodipine Intravenous Injection (Nimotop®, Bayer) and Digoxin Elixir Pediatric (Lanoxin®, GSK) are examples of co-solvent formulations [67,68].
(9)Miceller solublization
The use of surfactants to improve the dissolution performance of poorly soluble drug products has also been successfully employed. Surfactants can lower surface tension and improve the dissolution of lipophilic drugs in aqueous medium. They can also be used to stabilise drug suspensions. When the concentration of surfactants exceeds their critical micelle concentration (CMC, which is in the range of 0.05-0.10% for most surfactants), micelle formation occurs, entrapping the drugs within the micelles [69]. This process is known as micellisation and generally results in enhanced solubility of poorly soluble drugs. Commonly used non-ionic surfactants include polysorbates, polyoxyethylated castor oil, polyoxyethylated glycerides, lauroyl macroglycerides and mono- and di-fatty acid esters of low molecular weight polyethylene glycols. Surfactants are also often used to stabilize microemulsions andsuspensions into which drugs are dissolved [70].Examples of poorly soluble compounds that use Micellar solubilization are antidiabetic drugs, gliclazide, glyburide, glimepiride, glipizide, repaglinide, pioglitazone and rosiglitazone [71]. Micelles are divided into 2 categories-
(a) Mixed micelles
As long chain phospholipids form bilayers when dispersed in water the preferred phase of short chain analogues is the micellar phase because it is related with chemical structure, temperature and water content. Mixed micelles have a hydrophobic core in which low soluble compounds can dissolve. A micellar solution is a thermodynamically stable system formed spontaneously in water and in organic solvents. Micelle formation can only occur above a certain solute concentration, the critical micellar concentration (CMC), and at solution temperatures above the critical micellar temperature (CMT), the small colloidal aggregates (micelles) are in rapid thermodynamic equilibrium with a measurable concentration of monomers. The size (mostly around 5 to 10 nm) and shape of micelles depend, ultimately on the chemical, structure of the detergent. The micelle solubilizes host molecules (i.e. drugs) in the micelle volume, but the penetration into the micelle depends over all on the inner space of the micelle, on the hydrophohicity of the drug and on the charge of the incorporated molecule. The interaction between micelles and lipophilic drugs leads to the formation of mixed micelles (MM), often called swollen micelles, too. The addition of salt, alcohol etc. can vary the degree of penetration into the micelle (co-solubilization). Toxic side effects of some tensides on human cells have to be considered beside bad taste. The kinetics of micelles is driven by both rapid micelle- monomer exchanges and by dissolution and new formation of micelles, but nevertheless the extent of water-amphiphile contact between water and methylene and methyl groups and an extreme disorder of the micelle interior but swollen micelles arc fluid systems, sufficiently stable to he used as delivery systems for stable drugs.
(b) Polymeric Micelles
Amphiphilic polymers assemble into nanoscopic supramolecular core-shell structures, known as polymeric micelles, which are under extensive study for drug delivery. Polymeric micelles may be safe for parenteral administration relative to existing solubilizing agents, permitting an increase in the dose of potent toxic and poorly water soluble compounds. Polymeric micelles solubilize important poorly water-soluble compounds, such as amphotericin B, propofol, paclitaxel, and photosensitizers. A major factor in drug solubilization is the compatibility of a drug and a core of a polymeric micelle. Pluronics, poly (ethylene glycol) (PEG)-phospholipid conjugates, PEG-b-poly (ester)s, and PEG-b-poly (L-amino acid)s considered for drug delivery. Polymeric micelles may circulate for prolonged periods in blood, evade host defenses, and gradually release drug and show a preferential accumulation at sites of disease such as solid tumors. Polymeric micelles inhibit p-glycoprotein at drug-resistant tumors, gastrointestinal tract, and blood/brain barrier, perhaps providing a way to overcome drug resistance in cancer and increase drug absorption from the gut, drug absorption into the brain, reduce the self-aggregation of polyene antibiotics, key membrane-acting drugs used to combat life threatening systemic fungal diseases. In this way, they may reduce its dose-limiting toxicity without a loss of antifungal activity.
(10)HYDROTROPHY
Hydrotrophy is a solubilisation process whereby addition of a large amount of second solute results in an increase in the aqueous solubility of another solute. Hydrotrophy designate the increase in solubility in water due to the presence of large amount of additives. The mechanism by which it improves solubility is more closely related to complexation involving a weak interaction between the hydrotrophic agents like sodium benzoate, sodium acetate, sodium alginate, urea and the poorly soluble drugs [72]. Solute consists of alkali metal salts of various organic acids. Hydrotropic agents are ionic organic salts. Additives or salts that increase solubility in given solvent are said to “salt in” the solute and those salts that decrease solubility “salt out” the solute. Several salts with large anions or cations that are themselves very soluble in water result in “salting in” of non electrolytes called “hydrotropic salts” a phenomenon known as “hydrotropism”. Hydrotropic solutions do not show colloidal properties and involve a weak interaction between the hydrotropic agent and solute. Advantages of Hydrotropic Solubilization Technique: because the solvent character is independent of pH, has high selectivity and does not requireemulsification. It only requires mixing the drug with the hydrotrope in water. It does not require chemical modification of hydrophobic drugs, use of organic solvents, or preparation of emulsion system [73].The classification of hydrotropes on the basis of molecular structure is difficult, since a wide variety of compounds have been reported to exhibit hydrotropic behaviour. Specific examples may include ethanol, aromatic alcohols like resorcinol, pyrogallol, catechol, aand b-naphthols and salicylates, alkaloids like caffeine and nicotine, ionic surfactants like diacids, SDS (sodium dodecyl sulphate) and dodecylated oxidibenzene [74]. The aromatic hydrotropes with anionic head groups are mostly studied compounds. They are large in number because of isomerism and their effective hydrotrope action may be due to the availability of interactive piorbitals. Hydrotropes with cationic hydrophilic group are rare, e.g. salts of aromatic amines, such as procaine hydrochloride. Besides enhancing the solubilization of compounds in water, they are known to exhibit influences on surfactant aggregation leading to micelle formation, phase manifestation of multicomponent systems with reference to nanodispersions and conductance percolation, clouding of surfactants and polymers, etc. Other techniques that enhance the solubility of poorly water soluble drugs include salt formation, change in dielectric constant of solvent, Chemical modification of the drug, use of hydrates or solvates, use of Soluble prodrug, Application of ultrasonic waves, spherical crystallization [75].
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
(11)SOLID DISPERSIONS
In this technique, a poorly soluble drug is dispersed in a highly soluble solid hydrophilic matrix, which enhances the dissolution of the drug. Solid dispersion techniques can yield eutectic (nonmolecular level mixing) or solid solution (molecular level mixing) products [76,77]. A solid dispersion of carbamazepine in polyethylene glycol 4000 (PEG-4000) increased the rate and extent of dissolution of carbamazepine. In this method, a precipitation vessel was loaded with solution of carbamazepine and PEG4000 in acetone, which was expanded with supercritical CO2 from the bottom of the vessel to obtain solvent-free particles. Eutectic dispersions are homogeneous dispersions of crystalline or amorphous drugs in crystalline or amorphous carriers. In the solid solution form, the drug could be partially or completely soluble in the dispersing matrix.A solid dispersion of griseofulvin and polyethylene glycol 8000 (Gris-PEG®) is commercially available. Presence of the drug in microcrystalline state, improved wettability and formation of high free energy amorphous forms of the drug during solid dispersion formation contribute towards enhancement of drug solubilisation. Despite the promising aspects of dissolution enhancement and simplicity of concept, the solid dispersion technique has failed to gain popularity due to manufacturing, stability and scale-up issues [78,79]. The concept of solid dispersions was originally proposed by Sekiguchi and Obi, who investigated the generation and dissolution performance of eutectic melts of a sulfonamide drug and a water-soluble carrier in the early 1960 [80]. Solid dispersions represent a useful pharmaceutical technique for increasing the dissolution, absorption and therapeutic efficacy of drugs in dosage forms. The most commonly used hydrophilic carriers for solid dispersions include polyvinylpyrrolidone, polyethylene glycols, Plasdone-S630, Tween-80, Docusate sodium, Myrj-52, Pluronic-F68 and Sodium Lauryl Sulphate used. The solubility of celecoxib, halofantrine1, ritonavir can be improved by solid dispersion using suitable hydrophilic carriers. There are various techniques (Table 2) to prepare the solid dispersion of hydrophobic drugs to improve their aqueous solubility.
Table 2. Various techniques to prepare the solid dispersion of hydrophobic drugs to improve their aqueous solubility
|
S.No. |
Name of techniques |
Detail of techniques
|
|
1. |
Hot melt method (fusion method) |
The physical mixture of a drug and a water-soluble carrier was heated directly until it melted. The melted mixture was then cooled and solidified rapidly in an ice bath under rigorous stirring. The final solid mass was crushed, pulverized, and sieved, which can be compressed into tablets with the help of tabletting agents. The melting point of a binary system is dependent upon its composition, i.e., the selection of the carrier and the weight fraction of the drug in the system [81]. |
|
2. |
Solvent Evaporation Method |
The first to dissolve both the drug and the carrier in a common solvent and then evaporate the solvent under vacuum to produce a solid solution. This enabled them to produce a solid solution of the highly lipophilic β-carotene in the highly water soluble carrier polyvinylpyrrolidone. Many investigators studied solid dispersion of meloxicam15, naproxen and nimesulide using solvent evaporation technique. |
|
3. |
Hot melt extrusion |
Hot melt extrusion is essentially the same as the fusion method except that intense mixing of the components is induced by the extruder. Just like in the traditional fusion process, miscibility of drug and matrix can be a problem. High shear forces resulting in high local temperature in the extruder is a problem for heat sensitive materials. |
(12)Nanosuspension
A pharmaceutical nanosuspension is biphasic systems consisting of nano sized drug particles stabilized by surfactants for either oral and topical use or parenteral and pulmonary administration. Nanosuspension technology has been developed as a promising candidate for efficient delivery of hydrophobic drugs [82]. This technology is applied to poorly soluble drugs that are insoluble in both water and oils. The particle size distribution of the solid particles in nanosuspensions is usually less than one micron with an average particle size ranging between 200 and 600 nm. There are various methods for preparation of nanosuspension include Media Milling (Nanocrystals), High Pressure Homogenization in water (Dissocubes), High Pressure Homogenization in nonaqueous media (Nanopure) and combination of Precipitation and High-Pressure Homogenization (Nanoedege) [83]. Some of the techniques are discussed in table 3.
Table 3.Various techniques to prepare the nanosuspension of hydrophobic drugs to improve their aqueous solubility
|
S.No. |
Name of techniques |
Detail of techniques
|
|
1. |
Precipitation Techniques |
In precipitation technique the drug is dissolved in a solvent, which is then added to non-solvent to precipitate the crystals. Nanosuspension of Danazol, Naproxen, prepared by precipitation technique to improve their dissolution rate and oral bioavailability. |
|
2. |
Media milling (Nanocrystals or Nanosystems) |
The nanosuspensions are prepared by using high-shear media mills. The milling chamber charged with milling media, water, drug and stabilizer is rotated at a very high shear rate under controlled temperatures for several days (at least 2-7 days). The milling medium is composed of glass, Zirconium oxide or highly cross-linked polystyrene resin. The high energy shear forces are generated as a result of the impaction of the milling media. |
(13)Floating Granules
Patel Rajanikant et al. [84] utilized a novel approach for dissolution enhancement of ibuprofen (a weekly acidic, non-steroidal anti inflammatory drug) by preparing floating formulation. Drug having high permeability through stomach because it remain 99.9 % unionize in stomach (pKa of Ibuprofen - 4.43, pH of gastric fluid - 1.2) and mostly permeable through stomach but due to its solubility limitation it can’t enter in to systemic circulation and gastric empting time is 30 min to 2 hr. After this time ibuprofen goes in to small intestine where it is solubilised but can’t permeate through its membrane. It was logically decided to design such formulations which retain in stomach for more than 2 hrs because drug was not completely soluble within 2 hrs hence to dissolve completely in stomach region, this can be achieved by making floating dosage form. Floating ibuprofen granules were prepared by fusion method. Ibuprofen (200 mg divided in to 50 mg and 150 mg), gelucire 44/14 (350 mg melted) and ibuprofen (50 mg) added, disperse with glass road for uniform distribution of drug in to molted carrier, remaining 150 mg ibuprofen added in to molted Gelucire 44/14, this whole dispersion added in to molted gelucire 43/01. In optimized formulation, Granules remain floated for 3 hrs., gave 100%drug release in 150 minute in stomach region where it remain in 99.9% unionize form and absorbed to systemic circulation.
(14)Cryogenic techniques
Cryogenic techniques have been developed to creating nanostructured amorphous drug particles with high degree of porosity at very low temperature conditions so enhance the dissolution rate of drugs. Cryogenic inventions can be defined by the type of injection device (capillary, rotary, pneumatic and ultrasonic nozzle), location of nozzle (above or under the liquid level) and the composition of cryogenic liquid (hydrofluoroalkanes, N2, Ar, O2 and organic solvents). After cryogenic processing, dry powder can be obtained by various drying processes (spray freeze drying, atmospheric freeze drying, vacuum freeze drying and lyophilisation). Different types of cryogenic techniques (table 4) were invented like:
Table 4. Different types of cryogenic techniques
|
S.No. |
Name of cryogenic techniques |
Detail of cryogenic technique |
|
1. |
Spray freezing onto cryogenic fluids |
The drug and the carrier (mannitol, maltose, lactose, inositol or dextran) were dissolved in water and atomized above the surface of a boiling agitated fluorocarbon refrigerant. Sonication probe can be placed in the stirred refrigerant to enhance the dispersion of aqueous solution. |
|
2. |
Spray freezing into cryogenic fluids (SFL) |
It incorporates direct liquid – liquid impingement between the automized feed solution and cyogenic liquid to provide more intense atomization into microdroplets and consequently significantly faster freezing rates. The frozen particles are then lyophilized to obtain dry. |
|
3. |
Spray freezing into vapor over liquid (SFV/L) |
Freezing of drugs solution in cryogenic fluid vapours and subsequent removal of frozen solvent produces fine drug particles with high wettability [85]. During SFV/L the atomized droplets typically start to freeze in the vapor phase before they contact the cryogenic liquid. As the solvent freezes, the drug becomes supersaturated in the unfrozen regions of the atomized droplet, so fine drug particles may nucleate and grow. |
|
4. |
Ultra-rapid freezing (URF) |
Ultra?rapid freezing is a novel cryogenic technology that creates nanostructured drug particles with greatly enhanced surface area and desired surface morphology by using solid cryogenic substances. Application of drugs solution to the solid surface of cryogenic substrate leading to instantaneous freezing and subsequent lyophilization for removal of solvent forms micronized drug powder with improved solubility. |
|
5. |
High pressure homogenization |
The suspension of a drug and surfactant is forced under pressure through a nanosized aperture valve of a high pressure homogenizer. The principle of this method is based on cavitation in the aqueous phase. The particles cavitations forces are sufficiently high to convert the drug microparticles into nanoparticles. |
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE
(15) Nanocrystallization
The nanocrystallization is defined as a way of diminishing drug particles to the size range of 1-1000 nanometers. There are two distinct methods used for producing nanocrystals; ’bottom-up’ and ’top-down’ development. The top-down methods (i.e. Milling and High pressure homogenization) start milling down from macroscopic level, e.g. from a powder that is micron sized. In bottom-up methods (i.e. Precipitation and Cryo-vacuum method), nanoscale materials are chemically composed from atomic and molecular components.
(15.1) Milling
Nanoscale particles can be produced by wet milling process. In ball mills, particle size reduction is achieved by using both impact and attrition forces. The most common models are a tumbling ball mill and a stirred media mill. The degradation of mill surfaces and subsequent suspension contamination are problems of this method.
(15.2) High pressure homogenization
In high pressure homogenization, an aqueous dispersion of the crystalline drug particles is passed with high pressure through a narrow homogenization gap with a very high velocity. Homogenization can be performed in water or alternatively in non-aqueous media or water-reduced media. The particles are disintegrated by cavitations and shear forces. The static pressure exerted on the liquid causes the liquid to boil forming gas bubbles. When exiting from the gap, gas bubbles collapse under normal air pressure. This produces shock waves which make the crystals collide, leading to particle disintegration. A heat exchanger should be used when operating on temperature sensitive materials because high pressure homogenization causes increase in the sample temperature .The particle size obtained during the homogenization process depends primarily on the nature of the drug, the pressure applied and the number of homogenization cycles.
(15.3)Precipitation
In the precipitation method, a dilute solution is first produced by dissolving the substance in a solvent. The solution with the drug is then injected into water, which acts as a bad solvent. At the time of injection, the water has to be stirred efficiently so that the substance will precipitate as nanocrystals. Nanocrystals can be removed from the solution by filtering and then dried in air.
(15.4) Cryo-vacuum method
In the cryo-vacuum method, the active ingredient is first dissolved in water to attain a quasi-saturated solution. The method is based on sudden cooling of a solvent by immersing the solution in liquid nitrogen (-196 ºC) which causes a very fast rise in the degree of saturation based on the decrease of solubility and development of ice crystals when the temperature drops below 0 ºC. This leads to a fast nucleation of the dissolved substance at the edges of the ice crystals. The solvent must be completely frozen before the vessel is removed from the liquid nitrogen. Next the solvent is removed by sublimation in a lyophilization chamber where the temperature is kept at constant -22 ºC and the pressure is lowered to 10-2 mbar. Cryo-assisted sublimation makes it possible to remove the solvent without changing the size and habit of the particles produced, so they will remain crystalline. The method yields very pure nanocrystals since there is no need to use surfactants or harmful reagents.
(15.5) Nanomorph
The Nanomorph technology is to convert drug substances with low water-solubility from a coarse crystalline state into amorphous nanoparticles. A suspension of drug substance in solvent is fed into a chamber, where it is rapidly mixed with another solvent. Immediately the drug substance suspension is converted into a true molecular solution. The admixture of an aqueous solution of a polymer induces precipitation of the drug substance. The polymer keeps the drug substance particles in their nanoparticulate state and prevents them from aggregation or growth. Water redispersable dry powders can be obtained from the nanosized dispersion by conventional methods, e.g. spray-drying. Using this technology the coarse crystalline drug substances are transformed into a nanodispersed amorphous state, without any physical milling or grinding procedures. It leads to the preparation of amorphous nanoparticles [86]. Table 5,6 shows that nanotechnology approaches to improve the solubility of hydrophobic drugs.
Table 5. Nanotechnology approaches to improve the solubility of hydrophobic drugs
|
S.No. |
Nanoparticulate technologies |
Description |
|
1. |
Nanocrystal
|
Nanocrystal drug particles (<1,000 nm) produced by wet-milling and stabilised against agglomeration through surface adsorption of stabilizers; applied to NMEs eg aprepitant/reformulation of existing drugs eg. Sirolimus. |
|
2. |
Biorise |
Nanocrystals/amorphous drug produced by physical breakdown of the crystal lattice and stabilised with biocompatible carriers (swellable microparticles or cyclodextrins). |
|
3. |
IDD (Insoluble Drug Delivery) |
Micro-nm particulate/droplet water-insoluble drug core stabilized by phospholipids; formulations are produced by high shear, cavitations or impaction. |
|
4. |
CAP (Calcium Phosphate-based nanoparticles) |
For improved oral bioavailability of hormones /proteins such as insulin; also as vaccine adjuvant. |
|
5. |
NAB (Nanoparticle Albumin-Bound technology) |
Injectable suspension of biocompatible protein with drug improves solubility/removes need for toxic solvents; eg paclitaxel-albumin nanoparticles, injectable suspension of biocompatible protein with drug improves. |
|
6. |
Nanoedge |
Nanoedge technology: drug particle size reduction to nanorange by platforms including direct homogenization, micro precipitation, lipid emulsions and other dispersed-phase technology. |
|
7. |
BioSilicon |
Drug particles are structured within the nano-width pores of biocompatible biosilicon microparticles, membranes or fibres; gives controlled release/improves solubility of hydrophobic drugs. |
|
8. |
NanoGate |
Silicon membrane with nano-width pores (10-100 nm) used as part of an implantable system for drug delivery and biofiltration. |
|
9. |
NLC8 (Nanostructured Lipid Carriers) |
Nanostructured lipid particle dispersions with solid contents produced by high-pressure homogenization; lipid-drug conjugate nanoparticles provide high-loading capacity for hydrophilic drugs for oral delivery. |
Table 6. List of poorly soluble drugs along with methods for increasing solubility
|
S. No. |
For increasing methods solubility |
Poorly soluble drugs |
|
1 |
Nanoparticulation |
Dolargin, Loperamide, Tubocurarine, Doxorubicin, Ibuprofen, Diazepam, Naproxen, Carbamazepine, Griseofulvin, Nifedipine, Phytosterol |
|
2 |
Nanosuspension |
Omeprazol, Domperidone, Zidovudine |
|
3 |
Inulin Glass |
Cyclosporin, Diazepam, Amoxacilline, Bacitracin, D9tetra hydro cannabinol |
|
4 |
Mixed micelle |
Adriamycin, Doxorubicin, Paclitaxel |
|
5 |
RESS |
Aspirin, Ibuprofen, Nifedipine, β-Estradiol, Lovastatin, Stigmasterol, Salicylic acid |
|
6 |
SAS |
Lysozyme, Trypsin |
|
7 |
PCA |
Insulin, Hydrocortisone |
|
8 |
GAS |
Dexamethasone |
CONCLUSIONS
The solubility of the drug is the factor that controls the formulation of the drug as well as therapeutic efficacy of the drug, hence the most critical factor in the formulation development. Dissolution of drug is the rate determining step for oral absorption of the poorly water soluble drugs and solubility is also the basic requirement for the formulation and development of different dosage form of different drugs. The various techniques described above alone or in combination can be used to enhance the solubility of the drug. Although all techniques mentioned above could enhance the solubility, the choice of the method will be based on its effectiveness as well as safety in terms of biocompatibility of the excipient used. For orally administered drugs solubility is one of the rate limiting parameter to achieve their desired concentration in systemic circulation for pharmacological response. Solubility can be enhanced by many techniques and number of folds increase in solubility. Because of solubility problem of many drugs the bioavailability of them gets affected and hence solubility enhancement becomes necessary. It is now possible that to increase the solubility of poorly soluble drugs with the help of various techniques as mentioned above.
REFERENCES
[1] Bittner B., Mountfield R.J. Intravenous administration of poorly soluble new drug entities in early drug discovery: the potential impact of formulation on pharmacokinetic parameters. Current Opin. Drug Discov. Develop. 2002; 5: 59–71.
[2] Bittner B., Mountfield R.J. Formulations and related activities for the oral administration of poorly watersoluble compounds in early discovery animal studies. Pharm. Ind. 2002; 64: 800–807.
[3] Yu L.X., Gatlin L., Amidon G.L. Predicting oral drug absorption. In: Amidon, G. L., Lee P. I., Topp E. M. editors. Transport Processes in Pharmaceutical Systems. New York: Marcel Dekker, Inc.; p. 377 409.
[4] Meyer, M, C. “Bioavailability of drugs and bioequivalence In: Encyclopedia of Pharmaceutical Technology” New York. Marcel Dekker Inc.; 1998; 2, 33-58.
[5] Shargil, L, & Yu, “Applied Biopharmaceuitcs” Appleton-Century Crofts, Norwalk, C.T, 1985; 2, 193-203.
[6] Martin, A, “Physical pharmacy,” Lippincott Williams & Wilkins, A. Walters Kluwer Co, Philadelphia, 2003; 5, 410-418.
[7] Carstensen, J, T, “Pharmaceutical Preformulation” Teelinomoc Publishing Co. Inc, 1998; 14-47.
[8] Brahmankar D.M. et al, Bio pharmaceutics and Pharmacokinetics 2009; 349-357.
[9] Rinaki E, Valsami G, and Macheras P; Quantitative Biopharmacuetics Classification System; the central role of dose/solubility ratio. Pharm. Res. 2003; 20:1917.
[10]Lindenberg M., Kopp S., Dressman J.B. Classification of orally administered drugs on the World Health Organization model list of essential medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm. 2004; 58(2):265-78.
[11] Wu C.Y., Benet L.Z. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005; 22(1):11-23.
[12] Alfred Martin PH.D. et al, (1993) Physical Pharmacy, 4th edi., B.I. Publication, Pvt. Ltd. P.P.- 212-250
[13] Aulton M.E., Pharmaceutics, (2002) The science of dosage form design, 2nd edition, Churchill Livingstone, London, , P.P.-113 – 138, 234 – 252.
[14] Jain A., Ran Y., Yalkowsky S.H. Effect of pH-Sodium Lauryl Sulfate combination on solubilization of PG- 300995 (an Anti-HIV Agent): a technical note. AAPS PharmSciTech. 2004; 5(3): 45-48.
[15] Graham H., McMorland M., Joanne D., Wayne K., Peggy L.E., James E.A., James H.K.K., David R.G., Kerri R. Effect of pH-adjustment of bupivacaine on onset and duration of epidural analgesia in parturients. 1986; 33(5): 537-541.
[16] Gennaro A.R. editors. Remington, the science and practice of pharmacy, 21st ed. Lippincott, Williams & Wilkins, 2005; 867-868.
[17] Fiese E.F, Hagen T.A. Preformulation. In: Lachman L, Liberman H.A, Kanig J.L, editors. The theory and practice of industrial pharmacy. 3rd ed. Bombay: Varghees Publication House, 1990; 171-196.
[18] Allen L.V, Popovich, N.G, Ansel H.C., Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems, Lippincott, Williams & Wilkins 2005; 100-101.
[19] Fiese E.F., Hagen T.A. Preformulation. In: Lachman L., Liberman H.A., Kanig J.L., editors. The theory and practice of industrial pharmacy. 3rd ed. Bombay: Varghees Publication House; 1990. p. 171-196.
[20] Gennaro A.R. editors. Remington; the science and practice of pharmacy, 21st ed. Lippincott: Williams & Wilkins; 2005. p. 867-868
[21] Allen L.V., Popovich, N.G., Ansel H.C. Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems. Lippincott: Williams & Wilkins; 2005. p. 100-101.
[22] Venkatesh S., Li J., Xu Y., Rao V., Anderson B. D. Intrinsic solubility estimation and pH-solubility behaviour of cosalane (NSC 658586), an extremely hydrophobic diprotic acid. Pharm. Res. 1996; 13(10): 1453-1459.
[23] Danielsson I, Lindman B. The definition of microemulsion. Colloids Surfaces. 1981; 3: 391-392.
[24] Jayne Lawrence M., Rees G.D. Microemulsion-based media as novel drug delivery systems. Adv. Drug Deliver. Rev. 2000; 45(1):89-121.
[25] Holm R, Porter CJH, Edward GA, Mullertz, A, Kristensen HG, Charman WN, Examination of oral absorption and lymphatic transport of halofantrine in a triple cannulated canine model after administration in self-microemulsifying drug delivery systems (SMEDDS) containing structured triglycerides. European Journal of Pharmaceutical Sciences 2003; 20, 91-97.
[26] Pouton CW. Lipid formulation for oral administration of drugs, non- emulsifying, selfemulsifying drug delivery systems. European Journal of Pharmaceutical Sciences 2000; 11, S93- S98.
[27] Pouton CW. Formulation of self-microemulsifying delivery system. Advance Drug Delivery Reviews, 1997; 25, 47-58.
[28] Gershanik T, Benita S. positively charged self emulsifying oil formulation for improving oral bioavailability of progesterone, journal of Pharmaceutical Development and Technology, 1996; 1(2), 147 157.
[29] Shah N.H, Carvajal M.T, Patel C.I, Infeld M.H, Malick A.W. Self-emulsifying drug delivery systems (SEDDS) with poly-glycolyzed glyceride for improving in vitro dissolution and oral absorption of lipophilic drugs. International Journal of pharmacy and Pharmaceutical sciences, 1994; 106, 15-23.
[30] Lieberman H.A., Rieger M.M., Banker G.S. Pharmaceutical dosage forms, Disperse systems. New York: Marcel Dekker, Inc.; 1998. p. 149 -181.
[31] Attwood D. Microemulsions. In Colloidal Drug Delivery Systems, Kreuter H. (ed.) New York: Marcel Decker, Inc.; 1994. p. 31-40.
[32] Mueller E.A, Kovarik J.M, Van Bree J.B, Tetzloff W, Grevel J, Kutz K, Improved dose linearity of cyclosporine pharmacokinetics from a microemulsion Formulation. Pharmaceutical Research 1994; 11(2), 301-304.
[33] Gershanik T., Benzeno S., Benita S. Interaction of the self-emulsifying lipid drug delivery system with mucosa of everted rat intestine as a function of surface charge and droplet size. Pharm. Res. 1998; 15: 863-869.
[34] Shah N.H., Carvajal M.T., Patel C.I., Infeld M.H., Malick A.W. Selfemulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int. J. Pharm. 1994; 106: 15-23.
[35] Mueller E.A., Kovarik J.M., Van Bree J.B., Tetzloff W., Grevel J., Kutz K. Improved dose linearity of cyclosporine pharmacokinetics from a microemulsion Formulation. Pharm Res. 1994; 11(2): 301-304.
[36] Gershkovich P., Hoffman A. Uptake of lipophilic drugs by plasma derived isolated chylomicrons: linear correlation with intestinal lymphatic bioavailability. Eur. J. Pharm. Sci. 2005; 26(5): 394-404.
[37] Foppoli A., Sangalli M.E., Maroni A., Gazzaniga A., Caura, M. R., Giordano, F. Polymorphism of NCX 4016, an NO-releasing derivative of acetylsalicylic acid. J. Pharm. Sci. 2004; 93: 521–531.
[38] Morissette S.L., Soukasene S., Levinson D., Cima M.J. P Natl. Acad. Sci. USA, 2003; 100(5): 2180-2184.
[39] Chaumeil J.C. Micronisation: a method of improving the bioavailability of poorly soluble drugs, Methods and Findings in Experimental and Clinical Pharmacology. 1998; 20: 211-215.
[40] Adam M Persky and Jeffrey A Hughes; Solutions and Solubility. http://www.cop.ufl.edu/safezone/prokai/ pa5100 /pha5110.htm
[41] Blagden N., Gavan P.T., York P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates, Adv. Drug Del. Rev. 2007; 59(30): 617-630.
[42] Chaumeil J C; Micronization: a method of improving the bioavailability of poorly soluble drugs, 1998; Apr; 20(3):211-5.
[43] Muller R.H., Peters K., Becker R., Kruss B. Nanosuspension for IV administration of poorly soluble drugs - stability during sterilization and long term storage. Proc Int. Symp. Control. Rel. Bioact. Mater. 1995; 22: 574-575.
[44] Phillips E.M., Stella V.J. Rapid expansion from supercritical solutions: application to pharmaceutical processes. Int. J. Pharm. 1993; 94: 1-10.
[45] Subramaniam B, Rajewski R A, Snavely K; Pharmaceutical processing with supercritical carbon dioxide. J. Pharm. Sci. 1997; 86: 885-890.
[46] Sunkara G., Kompella U.B. Drug delivery applications of supercritical fluid technology. Drug.Del. Technol. 2002; 2: 44-50.
[47] Dohrn R., Bertakis E., Behrend O., Voutsas E., Tassios D. Melting point depression by using supercritical CO2 for a novel melt dipersion micronization process. J. Mole. Liq. 2007; 131-132.
[48] Loftsson T., Brewster M.E. Pharmaceutical applications of cyclodextrins: drug solubilization and stabilization. J. Pharm. Sci. 1996; 85: 1017-1025.
[49] Uekama K, Hirayama F, and Irie T; Cyclodextrin Drug Carrier Systems, Chem. Rev.,1998, 98, 2045 -2076.
[50] Loftsson T, Brewster M.E., Pharmaceutical applications of cyclodextrins and Drug solubilization and stabilization, Journal of pharmaceutical sciences, 1996; 85, 1017-25.
[51] Koester L.S., Bertuol J.B., Groch K.R., Xavier C.R., Moellerke R., Mayorga P., Dalla Costa T., Bassani V.L. Bioavailability of carbamazepine: betacyclodextrin complex in beagle dogs from hydroxypropylmethylcellulose matrix tablets, Eur. J. Pharm. Sci., 2004; 22(2-3): 201-207.
[52] Wen X, Tan F, Jing Z, Iiu Z. Prepration and study of the 1:2 Inclusion Complex of Carvedilol with β – Cyclodextrin, Simeoni, Journal of Pharmaceutical and Biomedical Analysis, 2004; 34, 517-523.
[53] Loftsson, T.; Masson, M. Cyclodextrins in topical drug formulations: theory and practice, International Journal of Pharmaceutics, 2001; 225, 15-30.
[54] Sato, K., Sugibayashi, K., Morimoto, Y. Species differences in percutaneous absorption of nicorandil, Journal of Pharmaceutical Sciences, 1991; 80, 104-7.
[55] Shaker, D.S., Ghanem, A.H., Li, S.K., Warner, K.S., Hashem, F.M., Higuchi, W.I. Mechanistic studies of the effect of hydroxypropyl-beta-cyclodextrin on in vitro transdermal permeation of corticosterone through hairless mouse skin, International journal of Pharmaceutics, 2003; 253, 1-11.
[56] Masson, Thorstein, Loftsoon, Gisli Masson, Einar Stefansson. Cyclodextrins as Permeation Enhancers; Some Theoretical Evaluations and invitro Testing. Journal of Control Release, 1997; 59,107-18.
[57] Rajewski R.A., Stella V.J. Pharmaceutical applications of cyclodextrins. in vivo drug delivery. J. Pharm. Sci. 1996; 85:1142-1169.
[58] Thompson D.O. Cyclodextrins, enabling excipients: their present and future use in pharmaceuticals. Critcal Reviews in Therapeutical Drug Carrier System, 1997; 14, 1-104.
[59] Baboota S, Bhaliwal M, Kohli K; Physicochemical Characterization, in-vitro Dissolution Behaviour, and Pharmacodynamic Studies of Reficoxib- Cyclodextrin Inclusion Compounds. Prepration and Properties of Reficoxib hydroxypropyl β- Cyclodextrin Inclusion Complex: a technical note. AAPS Pharm. Sci. Tech. 6(1) Article 14: 2005; E 83-E 89.
[60] Parikh R K, Mansuri N S, Gohel M C, Sonlwala M M; Dissolution enhancement of Nimesulide Using Complexation and Salt Formation Techniques. Indian Drugs. 2005; 42:149-53.
[61] Tsinontides S C, Rajnaik P, Pham D, Hunke W A, Placek J, Reynolds S D; Freeze drying-Principles and Practice for Sucessful Scale up to Manufacturing. Int. J. Pharm. 28(1): 2004:1-16.
[62] Tirucherai G S, Mitra A K; Effect of hydroxypropyl beta cyclodextrin complexation on aqueous solubility, stability, and corneal permeation of acyl ester prodrugs of ganciclovier. AAPS Pharm. Sci. Tech.4:2003: E45.
[63] Strickley, R.G.bSolubilizing excipients in oral and injectable formulations. Pharm. Res. 2004; 21: 201–230.
[64] Nema S., Washkuhn R.J., Brendel R.J. Excipients and their use in injectable products. Pharm. Sci. Technol. 1997; 51: 166-171.
[65] Rubino J.T., Yalkowsky S.H. Cosolvency and deviations from loglinear solubilization. Pharm. Res. 1987; 4: 231-236.
[66] Zhao L, Li P, Yalkowsky SH. Solubilization of fluasterone, Journal of Pharmaceutical Science, 1999; 88, 967–969.
[67] Seethala R., Fernandes, P.B. Handbook of Drug Screening. New York: Marcel Dekker, Inc.; 2001. p. 597-601
[68] Seedher N., Bhatia S. Solubility enhancement of Cox-2 inhibitors using various solvent systems. AAPS Pharm. Sci. Tech. 2003; 4: 33.
[69] Dutt G.B. Rotational diffusion of hydrophobic probes in Brij-35 micelles: effect of temperature on miceller internal environment. J. Phys. Chem. B. 2003; 107: 10546-10551.
[70] Yua B. G., Okanob T., Kataokac K., Kwona G. Polymeric micelles for drug delivery: solubilization and haemolytic activity of amphotericin B. Journal of Control. Rel. 1998; 53: 131-136.
[71] C.H. Hsu, Z. Cui, R. J. Mumper and M. Jay, Micellar Solubilization of Some Poorly Soluble Antidiabetic Drugs, AAPS PharmSciTech, 2008; 9 (2), 939-943.
[72] Cao FT, Guo J, Ping Q. The Physicochemical Characteristics of Freeze-Dried Scutellarin- Cyclodextrin Tetracomponent Complexes. Drug Dev. Ind. Pharm, 2005; 31,747-56.
[73] Balasubramanian, D. and Friberg, S. E., In Surface and Colloid Science (ed. Matijevic, E.), Plenum Press, New York, 1993, 15, 197-220.
[74] Roy, B. K. and Moulik, S. P., Colloids Surface. A Physicochemical Engineering Aspects, 2002; 203, 155–166.
[75] Patil S.V., Sahoo S. K., Pharmaceutical overview of spherical crystallization, Der Pharmacia Lettre, 2010; 2(1), 421-426.
[76] Craig D.Q.M. The mechanisms of drug release from solid dispersions in water soluble polymers. Int. J. Pharm. 2002; 203: 131-144.
[77] Sekiguchi K., Obi N. Studies on absorption of eutectic mixture-I: A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem. Pharm. Bull. 1961; 9: 866-872.
[78] Khoo S.M., Porter C.J.H., Charman W.N. The formulation of halofantrine as either nonsolubilising PEG 6000 or solubilising lipid based solid dispersions: physical stability and absolute bioavailability assessment. Int. J. Pharm., 2000; 205: 65-78.
[79] Hirasawa N., Ishise S., Miyata H., Danjo K. Application of nilvadipine solid dispersion to tablet formulation and manufacturing using crospovidone and methylcellulose as dispersion carriers. Chem. Pharm. Bull, 2004; 52: 244-247.
[80] Sekiguchi K, Obi N; Studies on absorption of eutectic mixtures. I.A. comparison of the behaviour of eutectic mixtures of sulphathiazole and that of ordinary sulphathiazole in man, Chem. Pharm. Bull 1961; 9: 866-872.
[81] Chiou W L, Riegelman S; Pharmaceutical Applications of Solid Dispersion Systems. J.Pharm Sci 1971; 60: 1281 1302.
[82] Nash R A; Suspensions. In: J Swarbrick, JC Boylan (ed). Encyclopedia of pharmaceutical technology. Second edition vol. 3. New York, Marcel dekker, 2002; 2045-3032.
[83] Chowdary K P R and Madhavi B L R, Novel drug delivery technologies for insoluble drugs. Ind.Drugs. 2005; 42(9): 557-563.
[84] Patel Rajanikant, Patel Nirav, Patel N M, Patel M M; A novel approach for dissolution enhancement of Ibuprofen by preparing floating granules, Int. J. Res. Pharm. Sci. Vol-1, Issue-1, 57-64, 2010.
[85] Buxton I R and Peach J M; Process and apparatus for freezing a liquid medium US4470202, 1984.
[86] NanoMorph technology for Amorphous Nanoparticles,<soliqs.com> [Accessed 05 Feb.]
NOW YOU CAN ALSO PUBLISH YOUR ARTICLE ONLINE.
SUBMIT YOUR ARTICLE/PROJECT AT articles@pharmatutor.org
Subscribe to PharmaTutor Alerts by Email
FIND OUT MORE ARTICLES AT OUR DATABASE








.png)

